4.1 PPI interaction with GABAA receptor
Comparative analysis of the binding ability of benzodiazepines and PPI to GABAA R showed that PPI binds to GABAA R in an almost similar fashion with high affinity and lower binding energy compared to benzodiazepines, and more hydrophobic interactions that increase the chances of easy binding. Therefore, it can be assumed that PPI may activate GABAA R, similar to benzodiazepines. Consequently, PPI may lead to neuronal degeneration, possibly causing dementia or AD (Table 1 and Figure 4).
4.2 PPI interaction with AMP-activated protein kinase
PPIs such as (R)-(+)-pantoprazole, esomeprazole, lansoprazole, (R)-omeprazole, (R)-tenatoprazole, and (S)-tenatoprazole bind to AMP-activated protein kinase (AMPK) with less energy compared to the known inhibitor (dorsomorphin) of AMPK. Interaction bonds contain the number of amino acids with hydrogen bonding and hydrophobic bonding. The data have shown the lowest level of energy in the binding site. Thus, PPI can be a potential inhibitor of AMPK, and its long-term use may keep inhibiting AMPK for the long term and thus may allow AD or dementia development (Table 2 and Figure 5).
| Complex with GABA(A) receptor | Energy (Kcal/mol) | Interaction bonds |
| Hydrogen bonding | Hydrophobic bonding |
| (R)-(+)-Pantoprazole | -8.1 | — | Thr87, Phe77, Arg65, Leu76, Tyr61, Phe62, Gly58 |
| Esomeprazole | -8.5 | — | Gly58, Thr87, Arg65, Tyr61, Leu76, Phe62 |
| Lansoprazole | -8.8 | — | Leu76, Tyr61, Arg65, Phe62, Gly58, Thr87 |
| (R)-Omeprazole | -8.5 | — | Phe62, Thr87, Leu76, Tyr61, Arg65, Gly58 |
| (R)-Tenatoprazole | -8.6 | — | Phe62, Gly58, Tyr61, Arg65, Leu76, Thr87 |
| (S)-Tenatoprazole | -8.6 | — | Gly58, Thr87, Leu76, Tyr61, Arg65, Phe62 |
| Benzodiazepine | -6.6 | Leu76 (2.91 A° & 2.88 A°) | Phe77, Thr87, Arg65, Tyr61 |
Table 1: Protein ligand interactions for 1GNU.
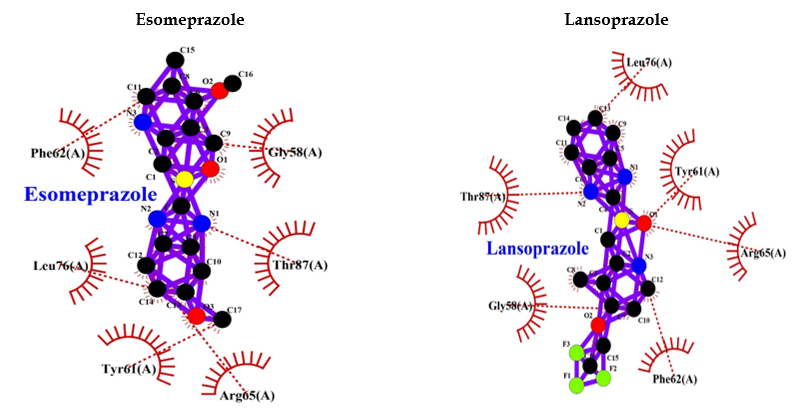
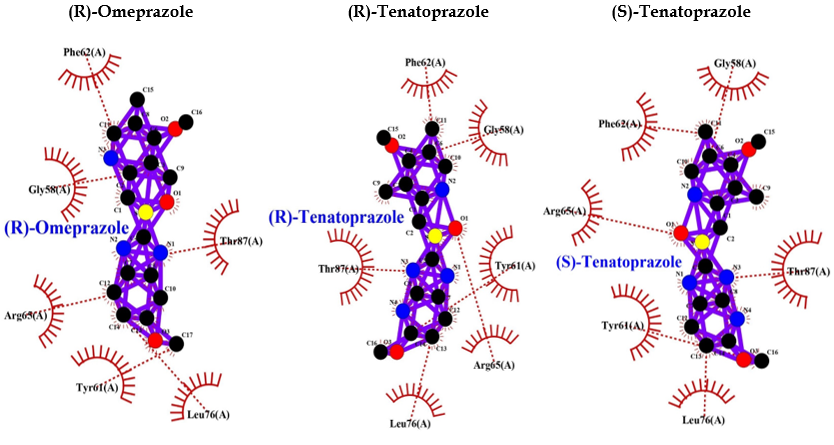
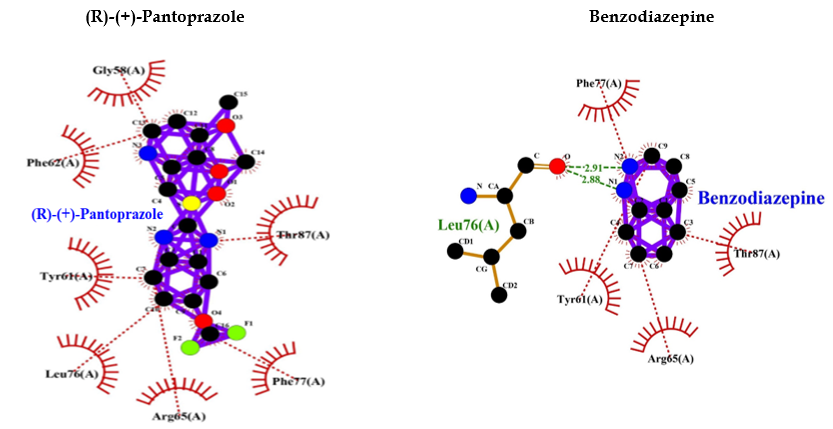 Figure 4: PPI Interaction with GABAA receptor.
Figure 4: PPI Interaction with GABAA receptor.
| Complex with AMPK | Energy (Kcal/mol) | Interaction bonds |
| Hydrogen bonding | Hydrophobic bonding |
| (R)-(+)-Pantoprazole | -8.1 | Val96 (3.12 A°) | Tyr95, Leu146, Glu94, Met93, Ile77, Leu22 |
| Esomeprazole | -8.1 | Val96 (3.15 A°) | Tyr95, Glu94, Met93, Ile77, Leu22 |
| Lansoprazole | -9.5 | Val96 (2.95 A°, 3.00 A°, 3.15 A°) | Tyr95, Glu94, Ile77, Met93, Leu22 |
| (R)-Omeprazole | -8.2 | — | Leu146, Val96, Leu22, Gly25, Ser161, Asn162, Lys45, Val30, Ala43, Ile77, Tyr95, Glu94 |
| (R)-Tenatoprazole | -8.3 | Val96 (3.17 A°) | Gly23, Asn162, Val30, Lys45, Glu94, Tyr95, Ile77, Gly25, Val24 |
| (S)-Tenatoprazole | -8.3 | Val96 (3.18 A°) | Gly25, Asn162, Lys45, Glu94, Ile77, Tyr95, Val30, Gly23, Val24 |
| Dorsomorphin | -1.1 | — | Pro213, Phe214, Val202 |
Table 2: Protein ligand interactions for 6BX6.
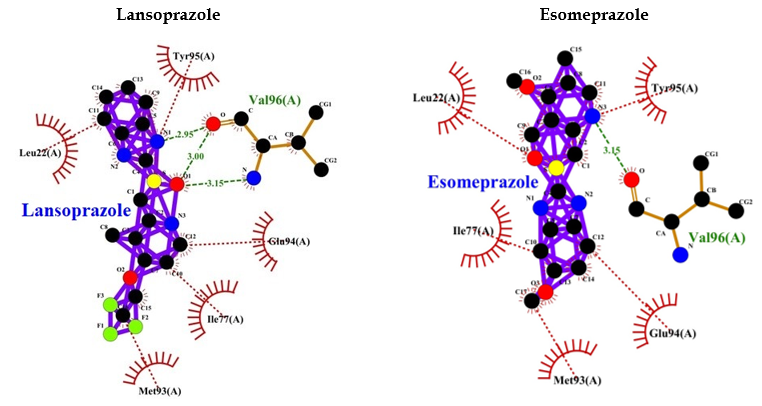
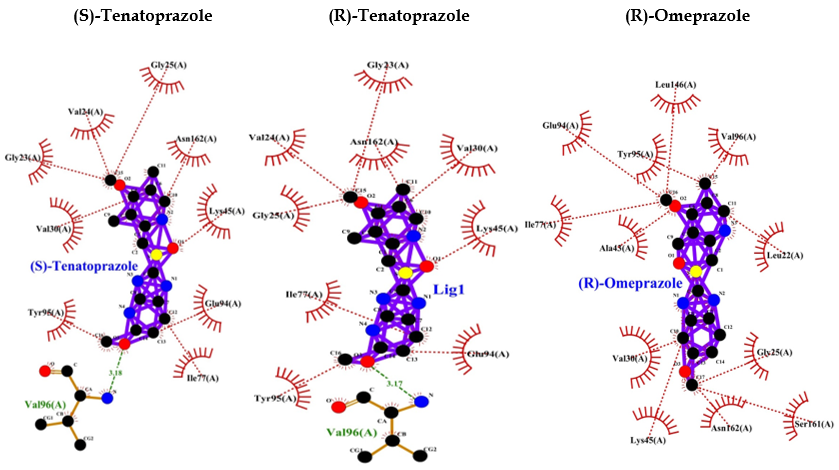
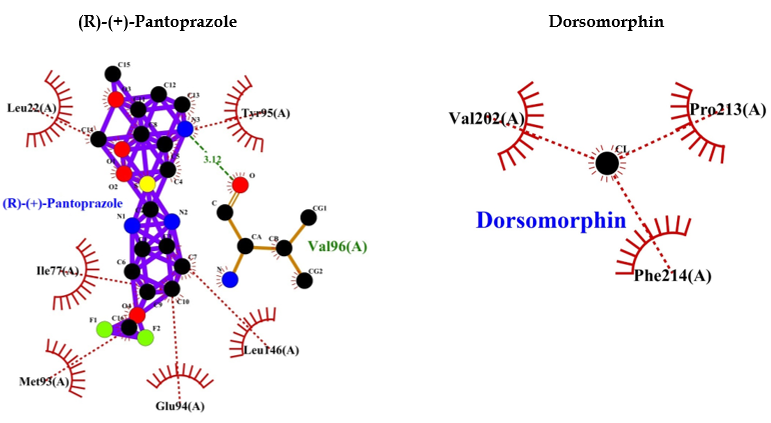
Figure 5: PPI interaction with AMPK.
4.3 PPI interaction with M1 muscarinic acetylcholine receptor
Benztropine is a known inhibitor of M1 muscarinic acetylcholine receptor (M1 mAChR). It shows a binding energy of -9.0 and a few hydrophobic bonds with no hydrogen bonds. Whereas, PPI shows interaction bonds of amino acids with hydrogen bonding and hydrophobic bonding, along with a binding energy lower than benztropine. These data indicate the lowest level of energy in the binding site, and thus, PPI can efficiently bind to M1 mAChR and inhibit it as benztropine (Table 3 and Figure 6).
| Complex with M1 muscarinic acetylcholine receptor | Energy (Kcal/mol) | Interaction bonds |
| Hydrogen bonding | Hydrophobic bonding |
| (R)-(+)-Pantoprazole | -9.5 | — | Thr192, Trp157, Tyr106, Tyr404, Ser109, Asp105, Cys407, Tyr408, Trp378, Asn382, Ala196, Phe197, Ala193 |
| Esomeprazole | -10.2 | — | Asn382, Phe197, Ala193, Ser109, Trp378, Tyr408, Cys407, Tyr404, Tyr106, Tyr381, Ala196, Thr192 |
| Lansoprazole | -10.6 | Tyr106 (3.11 A°) | Asn382, Tyr381, Trp378, Ser109, Cys407, Tyr408, Tyr404, Asp105 |
| (R)-Omeprazole | -9.6 | — | Asn422, Thr366, Asn60, Phe63, Ile119, Leu64, Ala363, Lys362 |
| (R)-Tenatoprazole | -9.5 | Asp122 (3.00 A°& 3.10 A°) | Glu360, Arg123, Phe63, Ile119, Asn60, Leu64, Val127, Ser126 |
| (S)-Tenatoprazole | -9.5 | Asp122 (2.99 A° & 3.10 A°) | Glu360, Arg123, Leu64, Phe63, Asn60, Ile119, Val127, Ser126 |
| Benztropine | -9.0 | — | Tyr404, Tyr85, Leu86, Glu401, Tyr82, Trp400 |
Table 3: Protein ligand interactions for 5CXV.
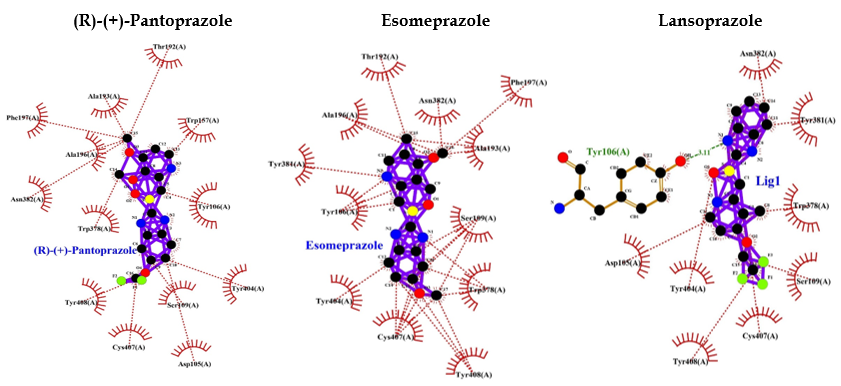
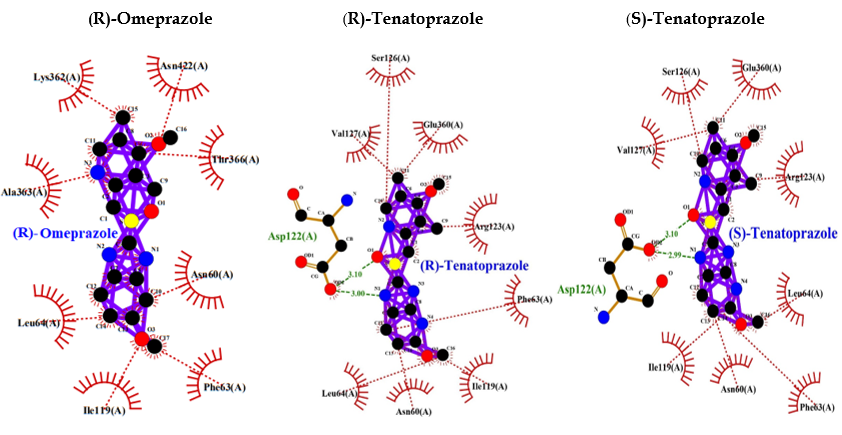
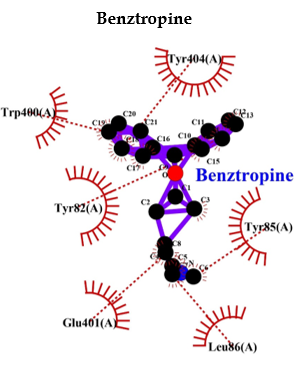
Figure 6: PPI Interaction with M1 muscarinic acetylcholine receptor (5CXV).
4.4 Postulated mechanism linking dementia and PPI use
The buildup of beta-amyloid has been implicated in the progression and pathogenesis of dementia syndromes such as AD in humans. Central nervous system (CNS) microglial cells use enzymes such as V-ATPase to degrade and scavenge beta-amyloid [2].
Murine models suggest that PPIs interfere with the activity of scavenger enzymes such as V-ATPase, can lead to the accumulation of beta-amyloid [9]. PPIs have also been associated with certain chronic kidney disease (CKD) that may also lead to cognitive impairment, dementia, or AD. Further studies are needed to elucidate the mechanisms linking PPIs with dementia in humans. In the current study, we have considered various CNS-associated proteins by previous studies, such as tau aggregation and beta-amyloid accumulation, to find what type of effects PPI may have on the CNS.
4.5 Effect of PPI on chronic kidney disease and eventually dementia
Kidney disease case reports have linked PPIs to acute interstitial nephritis (AIN) and acute kidney injury (AKI) since 1992. In 2016, two studies received widespread attention because they connected PPIs to an excess risk for CKD [10, 11]. Data retrieved retrospectively from a cohort of 10,482 patients who were actively followed and another larger cohort of 249,751 patients, it was found that PPIs were associated with a 50% increased risk in the smaller cohort and a 17% increased risk for CKD in the larger cohort.
Another study compared 173,321 PPI users with 20,270 H2 receptor antagonist (H2RA) users in a VA dataset [11]. They included patients who had a normal estimated glomerular filtration rate (eGFR) at baseline and followed patients for up to 5 years, and found a 1.8% absolute annual excess risk for CKD in PPI users compared to H2RA users.
These studies are thought-provoking since PPIs’ effect on the kidney may vary with the degree of severity within important comorbidity categories such as diabetes [9].
Evidence also exists that CKD is a risk factor for cognitive decline. Earlier studies explain that kidney disorder may be an important mechanism leading to cognitive impairment [12]. Cognitive dysfunction is a well-known complication of CKD [13]. AKI patients have a 3-fold higher risk of developing dementia compared with those without AKI. The association of AKI with dementia or death is valid in several studies and shows an increased risk of 60% [14].
How AKI may lead to cognitive dysfunction is unclear, but increased inflammation, oxidative stress, and endothelial dysfunction are all described complications of AKI [15–17]. In mouse models, ischemic AKI resulted in inflammation and functional changes in the brain. Specifically, compared with sham mice, those with AKI showed increased neuronal pyknosis and microgliosis in the brain [17]. In addition, the mice with AKI had significant microvascular dysfunction in the brain. Whether these changes occur in humans has not been determined, and further study is required. AKI is associated with long-term adverse outcomes. A significantly increased risk of dementia following AKI has been reported in patients without a previous history of cognitive dysfunction [13]. In a previous study conducted in Taiwan involving 2,905 patients, those with AKI had a greater risk of subsequently developing dementia than those without AKI, independent of cardiovascular risk factors [18]. PPI use is associated with an increased risk of AKI, incident CKD, and progression to end-stage renal disease (ESRD), ultimately leading to cognitive impairment [19].
4.6 The effect of PPIs on the central nervous system
AD or dementia is caused by the deposit of beta-amyloid and hyperphosphorylated tau protein in the brain of the patients, while in frontotemporal lobar degeneration, deposits of tau or TDP-43 can be characterized as Lewy body dementia, which is characterized by the presence of alpha-synuclein deposits [20–23].
Symptoms may be similar across different cognitive disorders in both the very early and late stages of the disease, making differential diagnosis challenging. However, the underlying causes of neurodegeneration differ in each condition. Evaluating the risk of PPIs on dementia and AD as a whole may provide essential insights into the disease. In this review, we discuss how PPIs may affect the CNS and contribute to neurodegeneration through various mechanisms.
4.7 PPI may inhibit ATP12A/ATP1AL1 (alpha polypeptide) gene product
The ATP12A/ATP1AL1 genes encode H+/K+-ATPase, which is expressed in the brain, colon, and placenta, while the ATP4A gene encodes H+/K+-ATPase in gastric epithelial cells. RNA blot analysis revealed that the colon had the highest levels of expression, whereas the kidney, uterus, heart, and forestomach had the lowest levels [109]. Moreover, this study has also shown the interaction of H+/K+-ATPase in gastric epithelial cells with rabeprazole and omeprazole [24, 25]. A few other isoforms of the H+/K+-ATPase are expressed in the CNS, which maintains acid-base and potassium homeostasis in neurons [24, 26]. V-ATPases are involved in both exocytosis and endocytosis in nerve terminals and are needed for the packing of neurotransmitters into synaptic vesicles by generating a proton gradient [27, 28]. PPIs, including omeprazole, lansoprazole, dexlansoprazole, rabeprazole, pantoprazole, and esomeprazole, bind to H+/K+-ATPases efficiently on the parietal cell membrane’s luminal surface and inhibit acid secretion [29, 30]. Most of the PPIs react with cysteine 813, though the site of reaction on the enzyme differs according to the type of PPI [30]. Due to the homology properties of P-type ATPases, PPIs can plausibly inhibit even other ionic pumps in the CNS and elsewhere. Hence, PPI may reduce pH in the brain, cerebrospinal fluid, and blood by inducing metabolic alterations.
According to the measurement of PPI passage through the blood-brain barrier (BBB), up to 15% of a single intravenous dose of omeprazole will pass through the BBB and enter the CNS [31]. Repetitive long-term use and 15% of this drug at each dose can potentially become a risk for the brain and can cause cognitive dysfunction. Lansoprazole has also been shown (in vitro and in vivo) to penetrate the BBB [32]. Lansoprazole, esomeprazole, and pantoprazole have been associated with headaches, dizziness, nervousness, tremor, sleep disturbances, and depression [33–35]. There have also been reports of senso-perceptual abnormalities (i.e., hallucinations) [36] and delirium [37] in very rare cases. Although the exact mechanisms of action of PPI on brain circuits and neurological side effects are not fully understood [38]. PPI drugs can facilitate tau and Aβ-induced neurotoxicity, which may increase AD progression and cognitive decline. Below, we discuss the most relevant physiopathological mechanisms.
4.8 PPIs and Aβ Plaques
Dementia build-up of β-amyloid (Aβ) protein predisposes to AD. Microglial cells use V-type ATPases to degrade amyloid-β, and PPIs may block V-ATPases to increase isoforms of amyloid-β in mice [39]. PPIs increase the development of Aβ plaques, which are one of the most well-known factors in the case of dementia [39]. Extracellular aggregation of Aβ plaques, which leads to oxidative and inflammatory damage in the brain, is one of the main hallmarks of AD. Aβ is produced through the proteolytic processing of a transmembrane protein, amyloid precursor protein (APP), by β-secretases (also known as β-site APP-cleaving enzyme 1 [BACE1]) and γ-secretases.
Amyloidogenic processing of APP is carried out by the sequential action of membrane-bound β- and γ-secretases. β-secretase cleaves APP into the membrane-tethered C-terminal fragments β (CTFβ or C99) and N-terminal sAPPβ. CTFβ is subsequently cleaved by γ-secretases into the extracellular Aβ and APP intracellular domain (AICD) [40]. Although the total number of Aβ plaques does not correlate well with AD severity, there is a direct effect on cognition and cell death in APP/tau transgenic mice because of neuronal loss and the astrocyte inflammatory response investigated the effect of PPIs on Aβ production using cell and animal models and suggested a novel hypothesis that considers PPIs as acting like inverse γ-secretase modulators (iGSMs), which change the γ-secretase cleavage site and thereby increase Aβ42 levels and decrease Aβ38 levels [39, 41]. PPIs also increase BACE1 activity, thereby increasing levels of Aβ37 and Aβ40. In AD, the major pathological species is thought to be Aβ42, but the most produced is Aβ40 [42]. PPIs and specifically lansoprazole was also noticed to alter the media pH responsible for amplifying the activity of other proteases, such as memprin-β, and generating Aβ2-37, Aβ2-40, and Aβ2-42 species. Moreover, Badiola et al. [39] were able to demonstrate that lansoprazole enhances Aβ production using in vivo and in vitro models, supporting the theory that PPIs affect AD by boosting Aβ production [39, 41]. PPIs inhibit vacuolar proton pumps (VPP) in microglia and macrophages, which acidifies lysosomes by pumping protons from the cytoplasm into the lumen of vacuoles [43, 44]. This acidic environment in lysosomes causes the degradation of fibrillary Aβ. As PPIs can cross the BBB, they act on V-ATPases in an inhibitory way, causing less degradation of fibrillary Aβ and hence a reduction in its clearance [31, 44]. To date, few studies have explained the relationship between the effects of PPIs and the presence of Aβ plaques. It would be interesting if future studies determine why Aβ plaque production increases or their clearance decreases with PPI use. Results from solid-state NMR measurements showed that amyloid fibril “cross β” structures are of two patterns: parallel and antiparallel. Tissue transglutaminase (tTG) causes crosslinking of Aβ peptides and indicates that the Aβ fibril is a hydrogen-bonded, parallel β-sheet with the propagation long axis of the Aβ fibril [45]. Similar to human AD cases, tTG was related to Aβ depositions in these AD models. Evidence for an early role of tTG in Aβ pathology was given in an earlier study [46].
One of the oxidative modifications involved in mediating Aβ toxicity through Aβ aggregation is the formation of dityrosine cross-links. Several studies have shown that Aβ can be converted to dityrosine through two different biochemical pathways. One method is peroxidase-catalyzed cross-linked tyrosine, and the second method is metal-catalyzed oxidative tyrosyl radical formation [47–50]. At certain concentrations, omeprazole induced HO-1, which also increased H2O2 levels [51, 52]. This increased hydrogen peroxide due to PPI use may cause the formation of dityrosine cross-links, which leads to the formation of Aβ aggregation that ultimately leads to cognitive impairment or dementia, or AD.
4.9 Role of PPI on Tau protein
A definitive diagnosis can only be confirmed histopathologically by the extensive presence of Aβ and neurofibrillary tangles (NFTs) in the neocortex of post-mortem brain tissue [53]. The main component of NFTs is paired helical filaments (PHFs) formed from hyperphosphorylated tau protein [54, 55]. Tau protein acts as a microtubule-associated protein in neuronal axons, stabilizing and inducing microtubule assembly [56]. When tau protein is hyperphosphorylated, it loses its ability to bind and stabilize microtubules, resulting in neuron degeneration [57]. According to the neuro-immunomodulation hypothesis of AD, the earliest CNS modifications before the clinical onset of AD are caused by a persistent inflammatory reaction, which causes excessive tau phosphorylation and triggers the development of PHFs and tau protein aggregates, eventually leading to cytoskeletal changes [58]. As a result, these lesions exist before the onset of clinical signs of AD [59]. They looked at over 2000 compounds to find agents for PET and discovered that quinoline and benzimidazole are high-affinity components of NFTs, and not senile plaques [59]. A docking experiment discovered significant hydrogen bond interactions between the NH group of lansoprazole’s benzimidazole ring and the tau core’s C-terminal hexapeptide (386TDHGAE391) [58]; lansoprazole has high lipophilicity and can cross the BBB within 37 min and can reach the brain; therefore, it has also been used as a radiotracer for PET imaging [60]. Tau undergoes multiple post-translational changes resulting in conformational modifications in aggregates that alter binding affinities and binding sites of tau protein [60]. Lansoprazole, indeed, with its high affinity for tau protein, can be used to create noninvasive techniques for diagnosing AD in the early stages. This has been proven that the tau protein effectively binds to PPI such as lansoprazole. Thus, the effect of other PPIs on the tau protein and its affinity may also increase aggregation and stabilize tau aggregates. It’s worth noting that TSP1 usually forms disulfide-linked trimers; it’s unclear if proteins with a proclivity for multimerization are more sensitive to omeprazole, but direct towards further investigation [61].
The appearance of abnormal phosphorylation of the microtubule-associated protein tau in the brains of patients with AD is a key characteristic of the disease’s development. Identification of the kinases involved in this mechanism, as well as the development of pharmacological agents to inhibit these molecules, has been a major focus of research. This analysis focuses on recent advances in tau phosphorylation’s physiological and pathological effects, as well as the role of phosphorylation in tau toxicity and pathological progression in AD. Therapeutic research is being reshaped by the emerging understanding of tau’s functions in cellular biology and the mechanisms by which phosphorylation controls tau activity [62].
The balance of tau kinase and phosphatase activities controls tau phosphorylation. This equilibrium has been proposed to be disrupted, which may lead to abnormal tau phosphorylation and hence, tau aggregation. Thus, identifying the potential causes of tau aggregate development and developing defense methods to deal with these lesions in AD necessitates a thorough understanding of tau dephosphorylating control modes. Stimulation of some tau phosphatases is one of the effective and reasonably precise treatments for reversing tau phosphorylation. We looked at tau protein phosphatases and analyzed their physiological functions and regulation, their function in tau phosphorylation, and their possible connection with AD in this article. We also reviewed the involvement of tau phosphatase, including protein phosphatase 2A (PP2A) [63].
4.10 Effect of PPI on GABAA receptor
Findings suggest that NR2A receptor activation is critical in limiting tau phosphorylation by the PKC/GSK3 pathway, and they support the concept that these receptors can function as a molecular device to prevent neuronal cell death and a variety of pathological conditions. After GABAA receptor (R) activation, tau phosphorylation at these residues was elicited by a pathway requiring cdk5, resulting in reduced PP2A interaction with tau [64]. A reduced PP2A will result in increased tau phosphorylation that may stabilize microtubules, leading to neuron degeneration. Thus, hyper-activation of GABAA R imbalances tau’s phosphorylation state, which may ultimately enhance the chances of dementia or AD via neuronal degeneration. According to previous positive interactive studies of PPI with some proteins, we made an interaction of GABAA R with various PPI and compared the interaction with a well-known GABAA R activator.
The binding of diazepam (benzodiazepines) to a specific allosteric site on GABAA R at the interface between α and γ subunits facilitates the inhibitory actions of GABA and can lead to a rapid increase in chloride/bicarbonate channels gating [65], which results in cumulative enhancement of GABA-mediated transmission at inhibitory synapses in the brain. Comparative analysis of the binding ability of benzodiazepines and PPI to GABAA R showed that it binds to GABAA R in an almost similar fashion with high affinity, and lower binding energy compared to benzodiazepines (Table 1 and Figure 4) and more hydrophobic interactions that increase the chances of easy binding. Therefore, it can be concluded that PPI may activate GABAA R as benzodiazepines. Accordingly, PPI may lead to neuronal degeneration, causing dementia or AD.
4.11 PPI as a potential inhibitor of AMP-activated protein kinase
A study also indicated that AMPK activation reduces tau phosphorylation, which improves brain function by inhibiting GSK3β in the AD-like model. These findings proved that AMPK might be a novel target for AD treatment in the future. Thus, activation of AMPK can be useful for preventing AD occurrence, and inhibition of AMPK will be unfavorable and may be associated with the development of AD [66]. In the current study, we performed an interactive study between AMPK and various PPIs, and our bioinformatics results showed that PPIs can inhibit AMPK, which may accelerate tau phosphorylation. This can be unfavorable for neuronal development due to imbalanced phosphorylation of tau and activation of GSK3β, which phosphorylates tau. The PPIs, such as (R)-(+)-pantoprazole, esomeprazole, lansoprazole, (R)-omeprazole, (R)-tenatoprazole, (S)-tenatoprazole, bind to AMPK with less energy compared to a known inhibitor (dorsomorphin) of AMPK [67]. Interaction bonds contain the number of amino acids with hydrogen bonding and hydrophobic bonding. The data has shown the lowest level of energy in the binding site. Thus, PPI can be a potential inhibitor of AMPK, and its chronic use may keep inhibiting AMPK for the long term and thus, may allow AD or dementia development (Table 2 and Figure 5).
4.12 PPI and acetylcholinesterase and M1 muscarinic acetylcholine receptor
A study also suggests that acetylcholinesterase (AChE) and the A-beta peptide may be involved in physiologically relevant interactions associated with the pathogenesis of AD [68]. An advanced in silico docking analysis followed by enzymological assessments was performed on PPIs against the core-cholinergic enzyme that is choline acetyltransferase (ChAT), which synthesizes acetylcholine (ACh). PPIs acted as inhibitors of ChAT, with high selectivity. Given that cholinergic dysfunction is a major driving force in dementia disorders [110]. This study mechanistically explains how prolonged PPI use may increase the incidence of dementia. Thus, prolonged PPI use in the elderly and patients with dementia or amyotrophic lateral sclerosis should be restricted.
4.13 Role of M1 muscarinic acetylcholine receptor in dementia and Alzheimer’s disease: PPI binds M1 mAChR in an inhibitory fashion
Aβ is an important player in AD and is derived from β-APP through sequential cleavages by β and γ-secretases: APP is cleaved by β-secretase to generate the large secreted derivative sAPPβ and the membrane-bound APP C-terminal fragment-β; the latter can be further cleaved by γ-secretase to generate Aβ and AICD. Alternatively, APP can be cleaved by α-secretase within the Aβ domain, which prevents Aβ production and instead generates secreted sAPPα, a neuroprotective protein [69, 70]. Interestingly, stimulation of M1 mAChR by agonists has been found to enhance sAPPα generation and reduce Aβ production [71, 72]. Stimulation of M1 mAChR is well-known to activate Protein kinase C (PKC). PKC is found to promote the activity of α-secretase [72] and the transfer of APP from the Golgi/trans-Golgi network to the cell surface [73]. M1 mAChR stimulation also activates ERK1/2, which modulates α-secretase activity and processing of APP [74], though some contradictory findings show opposite results [72]. In mouse AD models, M1 mAChR promotes brain Aβ plaque pathology by increasing amyloidogenic APP processing in neurons and the brain. M1 mAChR also affects BACE1, the rate-limiting enzyme for Aβ generation [75, 76]. APP/PS1/tau triple transgenic (3×Tg) AD mice were treated with AF267B, a selective M1 mAChR agonist. It reduces BACE1 endogenous level, accompanied by a decreased Aβ level via an unclear mechanism, directly interacts with BACE1, and mediates its proteasomal degradation [77, 78]. However, another study found that stimulation of M1 mAChR upregulates BACE1 levels in SK-SH-SY5Y cells via the PKC and MAPK signaling cascades [79]. M1 mAChR was found to induce the Wnt signaling pathway to counteract Aβ-induced neurotoxicity [80]. The involvement of M1 mAChR in AD is also manifested by its amelioration of tau pathology. Carbachol and AF102B (agonists) stimulate M1 mAChR in two time- and dose-dependent manners and decrease tau phosphorylation in PC12 cells [81]. AF267B (M1 mAChR agonist) lessens tau pathology by activating PKC and inhibiting GSK-3β in 3×Tg AD mice [77, 82]. Activation of M1 mAChR protects against apoptotic factors (such as DNA damage, oxidative stress, caspase activation, and mitochondrial impairment) in human neuroblastoma (SH-SY5Y cells) [83]. M1 mAChR cascade counteracts decreased cerebral blood flow, which is a pathological characteristic in AD, ischemic brain injury, and cognitive dysfunction [84, 85]. Uncoupling of M1 mAChR from G-protein in the hippocampal area, which is the most affected by Aβ, was reported in the postmortem brains of AD patients [86–90]. Aβ causes the uncoupling of M1 mAChR from G-protein, which inhibits the function of M1 mAChR [91, 92].
Eventually, these studies depicted that a decreased M1 mAChR signal transduction will reduce levels of sAPPα, thereby increasing Aβ, thus triggering the onset of pathological features of AD. Though the mechanism of Aβ disrupting mAChR-G-protein coupling is unclear and is palliated, implicating antioxidants and reducing the involvement of free radicals [91]. Ultimately, we found that inhibition of M1 mAChR results in dementia and AD, and since PPI has a wide range of interactions with various proteins, we selected to analyze the interaction between PPI and M1 mAChR. Benztropine is a well-known inhibitor of ACh muscarinic M1 and M3 receptors (mAChR). The implication of benztropine promotes differentiation of oligodendrocyte precursor cells and allows greater axonal remyelination in comparison to other drugs or molecules employed for treating multiple sclerosis [93]. More hydrophobic interactions were noticed in the case of PPI in comparison to benztropine, which suggests better chances of binding. PPI showed lower binding energy and higher chances of binding; lansoprazole and tenatoprazole also showed a hydrogen bond, which requires less binding energy. Whereas benztropine showed no hydrogen bonding, and thus PPIs have higher chances of binding, or a similar fashion of binding as benztropine.
PPI may also similarly inhibit M1 mAChR or potentially as benztropine (Table 3 and Figure 6).
4.14 PPIs and vitamin B12 deficiency
Gastric acidity is necessary for the absorption of vitamin B12, which is an essential water-soluble vitamin, obtained from different dietary sources such as fish, meat, dairy products, and fortified cereal [94]. The risk of B12 deficiency increases with age [95]. B12 is firmly bound to salivary R proteins and consequently requires acid-activated proteolytic digestion. PPI causes hypochlorhydria, resulting in vitamin B12 malabsorption [96]. PPI treatment for 2 years or longer showed a statistically significant association with an increased risk of B12 deficiency [97]. Whereas another study, in contrast, reported that a 3-year or longer PPI use had no change in B12 levels [98].
In recent studies, dementia and cognitive impairment have been associated with vitamin B12 deficiency due to chronic use of PPI [99]. Vitamin B12 is required for the production of nucleotides, phospholipids, and certain monoamine neurotransmitters [100]. Usually, vitamin B12 is responsible for converting tetrahydrofolate into methylcobalamin, which presents its methyl group to homocysteine, which is acted upon by methionine synthase and finally turns into methionine [99]. Vitamin B12 deficiency results in hyperhomocysteinemia and is considered a risk factor for cognitive impairment, dementia, and brain atrophy [101]. PP2A plays a crucial role in brain-associated disorders as it is the key serine/threonine phosphatase and prevents tau hyperphosphorylation in the brain [102]. Reduced methylation reduces PP2A function and leads to hyperphosphorylation and tau aggregation [99]. Hyperhomocysteinemia also increases Aβ production, while folate/vitamin B12 supplementation may attenuate these effects in animal models [103, 104].
According to these studies, elevated homocysteine levels are a strong risk factor for developing AD [105]. Alternatively, B12 can interact with thiol groups, i.e., cobalamin can directly bind to tau protein via cysteine, forming a B12/tau protein complex that prevents fibrillation of tau protein [99]. Vitamin B12 capping on cysteine also prevents tau aggregation. In summary, PPI causes vitamin B12 deficiency and hyperhomocysteinemia, leading to PP2A inactivation and tau hyperphosphorylation, which may result in cognitive impairment. The direct binding of vitamin B12 to tau protein, resulting in inhibited fibrillation and aggregation, is also one of the major causes [99].