Abstract
Alzheimer’s disease (AD) is a debilitating neurological disorder that causes cognitive and behavioral impairments. Because of the blood-brain barrier (BBB), medication cannot enter the brain, making the treatment of AD difficult. There are currently available therapeutic modalities that provide symptomatic relief, but they are also potentially unsafe. It has been found that phytoconstituents possess neuroprotective effects and the ability to target several pathogenic pathways associated with AD. Despite this, because of low bioavailability, poor solubility, and low BBB permeability, they have been unable to slow disease progression and suppress Alzheimer’s progression. Despite obstacles, nanotechnology is proving to be an effective tool for the delivery of brain drugs. In addition to reducing barriers, phytochemical-loaded nanocarriers can increase neuroprotective properties. Endocytosis is the most common mechanism of nanoparticles (NPs) transportation, including pinocytosis, receptor-mediated endocytosis, and phagocytosis. Drug delivery to the target site is accomplished through diffusion, degradation, or erosion. Many types of NPs are used, including dendrimers, liposomes, polymeric NPs, and microemulsions. The review provides an overview of prospective target molecules, delivery methodologies, phytoconstituents, and nanocarriers developed for treating and managing AD. Researchers seeking an alternative method to treat AD gained new insight by emphasizing prospects and obstacles.
Keywords
Alzheimer, targeted delivery, blood-brain barrier, nanotechnology, nanocapsule
Abbreviations
AD: Alzheimer’s disease; BBB: blood-brain barrier; NPs: nanoparticles; HIV: human immunodeficiency virus; AChE: acetylcholinesterase; FDA: Food and Drug Administration; CNS: central nervous system; APP: amyloid precursor protein; PS: presenilin; ChAT: choline acetyltransferase; APOE: apolipoprotein E; PA2: phospholipase A2; ILs: interleukins; TNF: tumor necrosis factor; Aβ: amyloid-β; ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3; BACE1: Beta-site amyloid precursor protein cleaving enzyme 1; PNPs: polymeric nanoparticles; PEG: polyethylene glycol; PAMAM: polyamidoamine; ATP: adenosine triphosphate; TPP-ceria NPs: triphenylphosphonium-conjugated ceria NPs; AGuIX NPs: gadolinium NPs; Kd: equilibrium dissociation constant; TTR: transthyretin; FAP: familial amyloid polyneuropathy; ATTR: amyloidosis TTR; AuNPs: gold NPs; SeNPs: selenium NPs; EGCG: epigallocatechin-3-gallate; NAA: N-acetylaspartate
Introduction
Alzheimer’s disease (AD) is a progressive and incurable disorder that affects cognition and behavior [1]. Due to Addison’s disease, there is a risk of plaques developing in the memory-making area of the cerebral cortex and other parts of the cerebral cortex used for thinking and making decisions [2]. It is estimated that 5.2 million Americans of all ages suffer from AD. AD is the sixth most common cause of death in the United States overall and the fifth most common among those 65 or older [3]. AD was responsible for 68% of deaths between 2000 and 2010, while heart, human immunodeficiency virus (HIV), and stroke disease were responsible for 16%, 42%, and 23% of deaths [4]. There is a strong likelihood that the number of people who will have AD by the year 2050 will almost triple from 5 million to an estimated 13.8 million unless advancements in medical science are made to prevent, delay, or stop the disease from progressing [5].
Present treatments have several limitations, and intranasal delivery seems to be one of the most promising ways to deliver drugs to the brain [6]. Drugs that modulate neurotransmitters or enzymes are available to treat cognitive impairments associated with AD. Treatments with acetylcholinesterase (AChE) inhibitors are most often discontinued due to adverse gastrointestinal effects such as nausea and vomiting [7]. The half-life of tacrine is short, requiring four doses a day to be effective [8]. Additionally, as a result of hepatotoxicity, patients taking medication were required to undergo periodic blood testing. The half-life of galantamine is 7 h, and that of rivastigmine is 2 h. There are some adverse effects associated with the use of memantine, such as constipation, dizziness, vomiting, and confusion [9].
It is common for therapeutic failure to be caused by adverse pharmacodynamics and pharmacokinetics [10]. The failure of pharmacotherapy is usually caused by tissue toxicity (kidney toxicity, hepatotoxicity, and neurotoxicity), inadequate physical chemistry (hydrophilicity), drug instability (photolysis, oxidation, and hydrolysis), a lack of absorption by biological membranes, and a lack of favorable pharmacokinetic parameters (such as plasma metabolism and intense) [11, 12]. Nanotechnology can enhance the pharmacodynamics and pharmacokinetics of medication and minimize its toxicity [13–16]. A key component in the development of nanomedicine is the controlled release of drugs into disease sites [17–19].
There are currently limitations to Food and Drug Administration (FDA)-approved AD drugs on the market, such as gastrointestinal side effects, high dosage regimes, ineffective brain targeting, and low bioavailability, which ultimately result in patients dissatisfied with the treatment and discontinuing it altogether [20, 21]. Nanotechnology plays a critical role in the process. This field has advanced in recent years, allowing therapeutic molecules to be delivered slowly across the blood-brain barrier (BBB) to reach the central nervous system (CNS) and removing other obstacles.
Pathophysiology of the Disease
AD can cause massive synaptic loss in several brain regions, including the cortex, the hippocampus, the entorhinal cortex, and the ventral striatum, which are involved in cognitive function [22]. Various senile plaques can be found in the brain parenchyma of Alzheimer’s patients, where fibrillar amyloid deposits can be found on the walls of blood vessels [23]. Plaques are caused by abnormal tau protein filament accumulation, neurofibrillary tangle formation, neuronal and synaptic loss, activation of glial cells, and inflammation [24]. Two hypotheses have been proposed regarding the pathophysiology and etiology of AD [25]. The first hypothesis is based on amyloid cascade neurodegeneration, while the second is based on cholinergic system dysfunction, including metal toxicity, tau aggregation, and inflammation [25].
The amyloid cascade neurodegenerative theory proposes that AD results from the cleavage of the amyloid precursor protein (APP) by proteolytic enzymes, leading to the production of amyloid plaques and the aggregation of amyloid-β (Aβ) molecules [26, 27]. Aβ deposition increases in AD patients with APP or presenilin (PS) mutations [28]. Deposition of Aβ is also associated with increased metal-mediated neurotoxic effects. As a result of high concentrations of Aβ, brain fibers are insoluble in water form [29]. There is a possibility that fibers are complexed with copper (Cu) and zinc (Zn), aggravating neuronal damage [30]. A study conducted in vitro showed that the Cu-Cu complexation resulted in neurotoxic hydrogen peroxide formation due to the Aβ-Cu complexation [31]. Moreover, it has been found that Zn, Cu, and iron are found in the amyloid deposits in the brain in patients with AD [32]. It may be possible to dissolve amyloid plaques in the postmortem tissues of patients who underwent AD using metal chelators [33]. In an in vivo study of an animal model of AD, chelating agents were also found to solubilize amyloid plaques [34].
Animal models of AD that have cholinergic dysfunction cause a memory deficit, according to the cholinergic hypothesis [35]. A study by Rossor et al. [36] and by Henke et al. [37] demonstrated degeneration of cholinergic neurons and reduced markers of cholinergic function in patients with AD. As a result, there was a reduction in the levels of choline acetyltransferase (ChAT) and a change in AChE activity in the cerebral cortex of patients with AD [37]. A study conducted by Soininen et al. [38] demonstrated that individuals with AD having the apolipoprotein E (APOE) ε4 allele exhibited a more severe deficit in cholinergic function than those without the APOE ε4 allele.
Phospholipase A2 (PA2) is an enzyme that synthesizes chemical mediators of inflammation and converts phosphatidylcholine into choline [39, 40]. However, PA2 has been reported to decrease AD patients’ frontal and parietal cortexes, reducing choline levels [41]. Due to the conversion of proteins ChAT and AChE, choline deficiency contributes to AD progression [39].
Tau protein is primarily responsible for promoting the association of tubulin monomers into microtubules, which ultimately modulate synaptic function and structure in neurons [42]. It is believed that tau protein phosphorylates abnormally in AD, causing the microtubules to disaggregate, accumulating inside the cells, and causing the cytoskeleton to become disorganized [43]. This results in a decline in dendritic or axonal transport in neurons because neurotrophic proteins cannot be transported intracellularly. There is an increase in reactive astrocytes in AD, and PA2 is highly expressed. Several proinflammatory molecules are released by astrocytes, including leukotrienes, interleukins (ILs), complement factors, prostaglandins, thromboxanes, proteases, and coagulation factors [44]. Alzheimer’s patients’ brains also contain abundant activated microglia cells [45]. The cells produce neurotoxic substances like nitric oxide, superoxide radicals, and glutamate. Following exposure to Aβ, microglia can release proinflammatory factors such as tumor necrosis factor (TNF), IL-1, IL-6, and IL-8 [46, 47].
Several complex mechanisms play a role in the progression and genesis of AD [48]. The understanding of molecular processes that regulate the cellular pathways involved in AD prognosis is improving rapidly due to recent advances in molecular biology. In turn, results may lead to identifying new molecular targets for treating and preventing AD [49].
Alzheimer's Disease Treatment Targets
AD is a complicated and mysterious condition [5]. Various hypotheses have been proposed to explain its pathogenesis, including the amyloid cascade and tau hyperphosphorylation hypotheses [50]. However, the exact cause of AD remains elusive. Several recent, widely studied targets for treating AD include oxidative stress, Aβ peptides, neurons, tau proteins, microenvironments, and microglial cells in the brain [51].
Toxic amyloid-β plaques and tau protein
There has long been a belief that the presence of neurofibrillary tangles formed by tau protein in the brain and Aβ plaques in the brain are the primary signs and symptoms of AD [52]. The amyloid cascade hypothesis holds that unbalanced production and removal of Aβ causes toxic Aβ oligomers to accumulate extracellularly. As a result, the amyloid cascade hypothesis triggers can induce fibrillation and lead to large deposits or plaques, resulting in synaptic dysfunction and neurodegeneration [26]. Further, a metal ion imbalance, specifically the dysregulation of Zn and Cu, promotes AD development by accelerating the deposition of Aβ plaques and generating reactive oxygen species (ROS). Addiction treatments such as metal-ion chelation have proven effective. Tau hyperphosphorylation is recognized to occur as a result of intracellular tau aggregation [53]. Normal neurons bind tau to microtubules, modulating intracellular trafficking with tubulin. The tau protein in AD is hyperphosphorylated, disrupting the function of microtubules [54]. Furthermore, toxic self-aggregated tau has a significant effect on synaptic function and neuronal function. Furthermore, there is growing evidence that the tau and Aβ pathologies synergistically accelerate AD progression [55]. For instance, Aβ aggregation is closely connected to the breakdown of normal tau into toxic forms. Therefore, co-targeting Aβ and tau pathology is expected to be more effective than treating each separately.
Microglia
The microglia, macrophages found throughout the CNS, are a promising therapeutic target for treating AD [56]. As part of their normal function, microglia phagocytose diseased cells and abnormal proteins and secrete anti-inflammatory cytokines and proinflammatory to maintain a stable brain environment [57]. AD, however, causes the microglia to become disordered, which makes them less able to process abnormal proteins. When excessive toxic agents are generated, abnormal brain conditions, such as inflammation, damage neurons and undermine microglia’s functions [58]. Therefore, AD conditions worsen significantly. Several studies have targeted microglia as a therapeutic strategy for AD, and the results have been more promising [59]. For instance, the signal transducer and activator of transcription 3 (STAT3) is crucial in inducing inflammatory mediator secretion. A decrease in STAT3 levels in dysfunctional microglia reduces inflammation and accelerates the clearance of Aβ [60]. Beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibition in microglia increases Aβ uptake and degradation in AD mice with superior therapeutic outcomes [61].
Neurons
Cholinergic neurons, in particular, play an essential role in forming memories [62]. Healthy neurons express several molecules to fight off attacks from inflammatory mediators. During AD progress, some neurons are damaged, and the formation of intracellular neurofibrillary tangles is the neurons’ most prominent pathological effect [63]. A dysfunctional tau pathway in neurons results in abnormal energy production and ROS, further activating microglia. Microglia activation leads to the release of inflammatory mediators, which further damage the neurons [64]. The dysfunction of neurons and microglia is thus influenced by each other [65]. Besides abnormal tau pathways in neurons, mitochondrial dysfunction occurs early in AD pathogenesis and plays a crucial role in accumulating Aβ and tau by causing oxidative stress [66]. So, it may be possible to delay AD progression by regulating dysfunctional neurons via therapeutic approaches, such as antioxidation combined with tau-targeting or neuroprotection coupled with mitochondrial targeting [67–69].
Oxidative stress environment
The presence of oxidative stress markers before the development of pathological damage has been demonstrated in research studies [70, 71]. Various studies have examined redox conditions’ contribution to AD progression by modulating the oxidative stress environment. An imbalance between ROS formation and clearance causes oxidative stress, which causes cellular damage. In addition to the AD-related pathways that act on ROS production, there are several ROS-related pathways [72]. For instance, Aβ accumulation accelerates ROS production. Additionally, mitochondrial dysfunction is strongly associated with oxidative stress. High ROS levels activate microglia and astrocytes, triggering a cascade of proinflammatory responses that further promote ROS production [73]. A high level of ROS also leads to synaptic dysfunction and neuronal degeneration. Creating a state of homeostasis for ROS in the brain has significantly impacted preventing and treating AD [74, 75].
Nanotechnology-Assisted Drug Delivery Strategies for Alzheimer's Disease
The current list of drugs approved to treat AD is all oral formulations, with rivastigmine being the only drug that also comes in a transdermal patch form [76]. Because most drugs lose their potency in the gastrointestinal tract and the hepatic system, a higher dose must be consumed to control disease progression or symptoms [77]. It is also necessary that the drug binds to serum albumin in the bloodstream for a satisfactory half-life before it can finally penetrate the BBB [78]. The consumption of dosages can lead to side effects like diarrhea and nausea, which can reduce the compatibility of the patients. Recent advances in nanotechnology have allowed nanoparticles (NPs) to overcome these hurdles. In particular, NPs can minimize side effects by reducing dosage and rapidly traversing the BBB to deliver the drug to a targeted area. To permeate the BBB, NPs must be between 1 and 100 nm in size [79].
As a result of formulated properties, they are target-specific, nontoxic, and biodegradable. Nanosystems in treating AD may allow for the effective transport and distribution of other neuroprotective molecules and drugs [80]. As a result of intranasal administration, the BBB is bypassed, and drugs can reach the brain directly. For the optimal effect of pharmacotherapy for patients with AD, nanodevices can be administered nasally, federally, or intravenously to target the brain as they traverse the BBB to improve their pharmacodynamic and bioavailability properties and decrease adverse effects [81]. Endocytosis is the most common mechanism of NP transportation, including pinocytosis, receptor-mediated endocytosis, and phagocytosis. Drug delivery to the target site is accomplished through diffusion, degradation, or erosion. Many types of NPs are used, including dendrimers, liposomes, polymeric NPs, and microemulsions.
Liposomes
The phospholipids are amphiphilic bilayers (that is, they can transport lipophilic and hydrophilic molecules) [82]. It has been proposed that some antibodies can inhibit tau pathology by phagocytosing the antibody-tau complex by microglial cells. They can also facilitate the clearance of lysosomal tau in neurons following endosomal uptake by nodes [83]. Several components are present in liposomes, including glycerophospholipids, phosphatidylcholine, and sphingomyelin. Cholesterol, which helps maintain liposome stability, is present in liposomes whose size is typically between 50 and 100 µm. Drugs are encapsulated inside lipid bubbles, which help prevent enzyme degradation and maintain efficacy [84].
Polymeric nanoparticles
The size of NPs is 1–100 nm. They are composed of natural or synthetic polymers [85]. Polymeric nanoparticles (PNPs) can be hydrophobic or hydrophilic depending on the type of part forming their outermost layer. They may be transported to the target site through receptor-mediated endocytosis or transcytosis of endothelial cells. Antibodies or polyethylene glycol (PEG) can enhance drug absorption when delivered via intranasal means, especially when PNPs are coated with antibodies. Poly (n-butyl cyanoacrylate) coated with polysorbate 80 enhanced drug delivery in AD cases when loaded with rivastigmine. In a model of experimental AD, antibodies decorated with NPs cure the memory defect completely [86]. Preparing NPs with distinct quantum properties can help diagnose and imaging conditions [87]. As polymeric NPs, they apply to nanogels, dendrimers, and nanocapsules in general [88].
Micro- and nanoemulsions
The NPs in this category are considered surfactant-based systems. The size ranges from 10–140 nm in microemulsions, while the size falls between 100 and 200 nm in nanoemulsions [89]. Typically, systems are described as oil-in-water heterogeneous systems, as oil is dispersed in water or another aqueous medium. An intranasal delivery of a nanoemulsion containing curcumin and resveratrol based on hyaluronic acid showed promising results. As part of the preparation process, spontaneous emulsification was used [77]. An alternative method of transmitting memantine may be using the nanoemulsion that has been produced [90] (Figure 1).
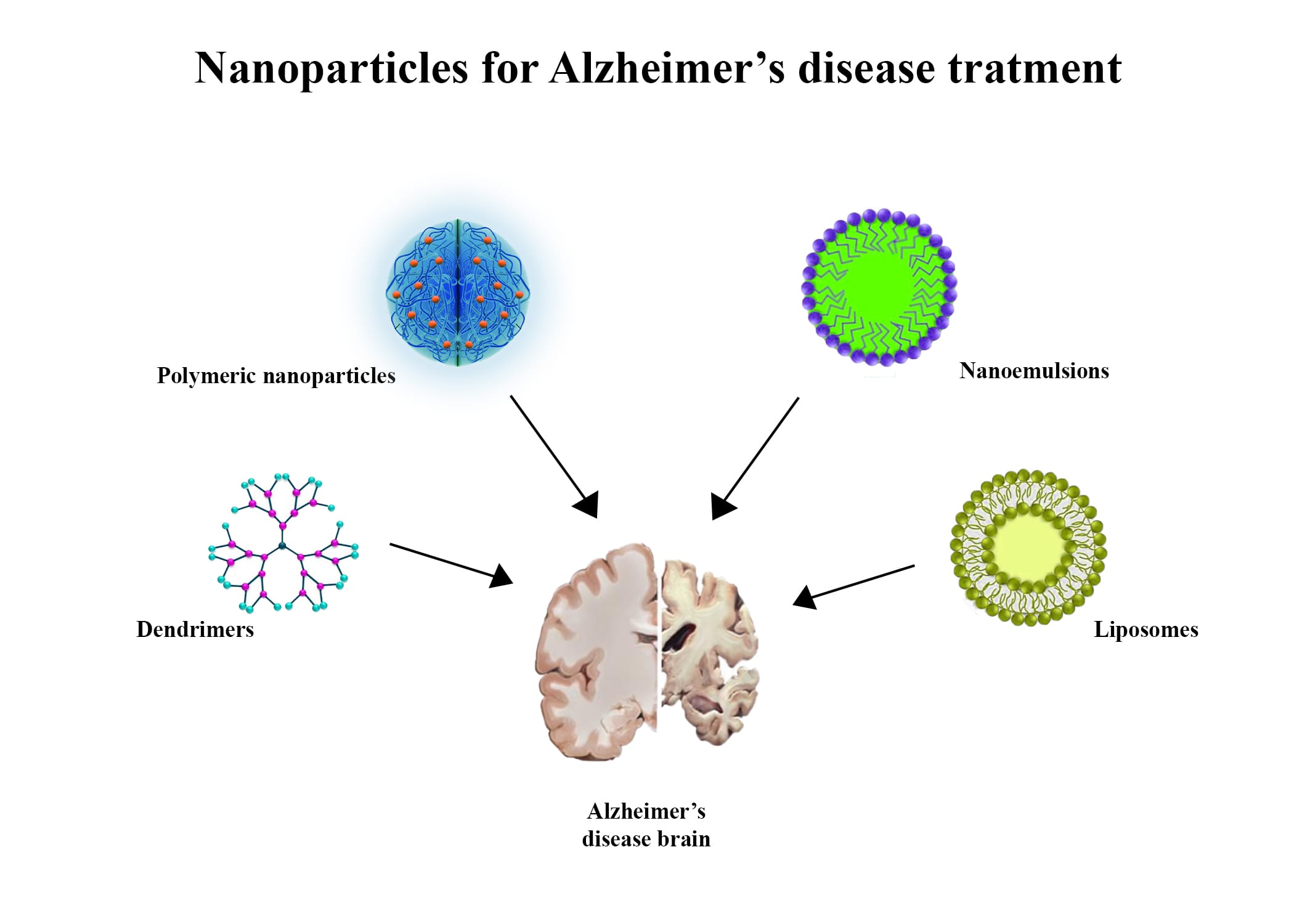 Figure 1: Four important nanoparticles and formulation for enhanced Alzheimer’s treatment.
Figure 1: Four important nanoparticles and formulation for enhanced Alzheimer’s treatment.
Dendrimers
In addition to cascade molecules, arborols are polymeric branched molecules with globular structures. Based on the structural nature of their ramification, they are similar to tree branches, which ramify while originating from a core. Convergent and divergent methods are both used to produce dendrimers. The inner cavity of the dendrimer and the outer surface has a high drug-loading capacity. Dendrimer sizes can be easily controlled by selecting the monomers and polymerization degree. Polyamidoamine (PAMAM) are the most commonly used dendrimers in drug delivery because they are hydrophilic, biocompatible, and nonimmunogenic. Dendrimers comprised groups of carboxyl and amines terminating hydrophobic ethylenediamine and methyl acrylate molecules. Dendrimers made from PAMAM are used in various applications, including drug delivery [91], diagnostic agents [92], gene transfection [93], and boron neutron capture therapy for patients with metastatic brain cancer (Figure 2).
 Figure 2: Dendrimer’s mechanism of action for Alzheimer’s therapy.
Figure 2: Dendrimer’s mechanism of action for Alzheimer’s therapy.
Metallic Nanoparticles in the Therapy of Alzheimer's Disease
Cerium-loaded nanoparticles in Alzheimer’s disease therapy
Research has revealed that ROS production and Aβ peptides interact with mitochondrial proteins, such as adenosine triphosphate (ATP) synthase, cyclophilin D, and alcohol dehydrogenase [94, 95]. Mitochondrial dysfunction precedes the build-up of AD plaques in the brain, a hallmark of the disease [96]. Therefore, therapeutic agents that can protect mitochondria from oxidative stress resulting from ROS can effectively prevent and treat AD at an early stage. It has been demonstrated that ceria (CeO2) NPs are capable of scavenging ROS by mimicking superoxide catalase and dismutase through their recyclable nature by reversible oxygen binding and switching between the Ce4+ (oxidized) and Ce3+ (reduced) states on their surface in a recycling manner [97, 98]. Various therapeutic effects have been demonstrated for ceria NPs, such as wound healing, neuroprotection, cardioprotection, and cancer and ocular disease treatment [99]. One example is Kwon et al. [100] production of triphenylphosphonium-conjugated ceria NPs (TPP-ceria NPs) that are localized to mitochondria and suppress neuronal death in a 5XFAD transgenic AD mouse model. Due to its lipophilicity, TPP can target mitochondria by exploiting the negative membrane potential of mitochondria. TPP-ceria NPs attenuated mouse reactive gliosis and mitochondrial morphological damage. TPP-ceria NPs have been shown to target mitochondria efficiently due to their excellent colloidal stability, small size (3 nm), hydrophobicity, small hydrodynamic diameter (22 nm), and positive zeta potential (+45 mV) [100].
Gadolinium nanoparticles in the diagnosis of Alzheimer’s disease
Gadolinium NPs (AGuIX NPs) were successfully attached to two peptides that contain the KLVFF sequence, which corresponds to the hydrophobic core of the Aβ peptide (residues 16-20), crucial for the formation of β-sheets [101, 102]. Using an atypical anti-parallel β-sheet motif, the Aβ fragment of the Aβ peptide binds to the full-length Aβ peptide [103]. The Aβ peptide also binds the peptide LPFFD’s hydrophobic region. According to plasmon resonance spectroscopy experiments, both functionalized NPs interacted with Aβ1–42 fibrils at equilibrium dissociation constant (Kd) values ranging between 350 and 403 micrograms, corresponding to the dose commonly used in clinical MRI [104]. It was found that these peptides did not interact with the V30M-TTR fibrils. It is believed that the fibrils that form amyloid are formed from the mutant form of transthyretin (TTR) associated with familial amyloid polyneuropathy (FAP), which is also referred to as hereditary amyloidosis related to TTR. Aβ peptide difference between the A and TTR proteins causes amyloidosis TTR (ATTR), a neurodegenerative disease characterized by fibrillation and misfolding of the protein [105]. A similar type of experiment was conducted with Pittsburgh compound B. Several have been reported to interact with Aβ1–42 fibrils and V30M-TTR fibrils, and the Kd values ranged from 6 mM to 10 mM, suggesting that this agent would be helpful in the diagnosis of amyloid vascular dementia and ATTR [106]. According to immunohistochemical studies, both AGuIX NPs exist in AD, such as neuropathologies in the mouse hippocampus. A high positive MRI contrast was associated with NPs, and they enhanced the MRI signal, improving image interpretation. Further, AGuIX NPs did not accumulate in specific organs as did iron oxide NPs. Therefore, their cytotoxicity was limited [107, 108].
Gold nanoparticles in the therapy and diagnosis of Alzheimer’s disease
The use of gold NPs (AuNPs) in treating AD has been compared to that of metallic NPs used to label and destroy cancer cells in an experimental technique [109]. AuNPs were conjugated to Aβ fibrils, and several days of incubation followed by several hours of exposure to weak microwave fields disintegrated the plaques. There was a much lower energy level in the fields than in conventional cell phones, which makes healthy cells unlikely to be affected. Irradiation caused the fibrils to disband for at least one week [110, 111]. As a result, the fibrils were broken up, and the reaggregation potential of the proteins was reduced. Liao et al. [112] reported that bare AuNPs inhibited Aβ fibrillation, resulting in fragmented spherical and fibril oligomers. As a result of the addition of AuNPs bare to preformed Aβ fibrils, however, ragged species were obtained, showing that the AuNPs had a preferential binding in the fibrillar structure. An incubation of positively charged AuNPs with carboxylate-coated AuNPs, but not with amine-conjugated AuNPs, reduced the neuroblastoma-mediated toxicity of Aβ. As a result of the study, AuNPs with negative surface potential may serve as nano-chaperones that inhibit and redirect pre-fibrillation, thus possibly serving as a power therapy for treating AD [112]. Neely et al. [113] conducted a study to examine an in vitro two-photon scattering assay based on monoclonal anti-tau antibodies-coated AuNPs, which showed promise for detecting AD tau proteins. In the presence of 20 ng/ml of tau protein, AuNPs coated with anti-tau antibody increased the intensity of two-photon Rayleigh scattering by approximately 16 times [113].
Ramassamy et al. [114], conducted a study to develop an ultrasensitive immunosensor built with AuNPs decorated with antibody fragments (Fab). The sensor explicitly recognizes Aβ complexes. A linear correlation between the peptide concentration and the result was observed over nine orders of magnitude. Compared to a system on a bare gold substrate, the detection limit was increased from 10 ng mL−1 to 1 fgmL−1, thanks to increased surface plasmon resonance.
Selenium nanoparticles in the therapy of Alzheimer’s disease
There is evidence that selenium and its redox cycles, including sodium selenite (IV), selenium (II), and sodium selenite (VI), effectively block ROS. Selenite and selenium NPs (SeNPs) have been shown to inhibit cytotoxicity and lower oxidative stress [115, 116]. Researchers have investigated how SeNPs inhibit Aβ aggregation extensively. Sialic acid was used to modify SeNPs, resulting in NPs that inhibited Aβ aggregation and crossed the BBB [94]. As well as modifying SeNPs with sialic acid and coating them with a highly permeabilized BBB peptide B6, epigallocatechin-3-gallate (EGCG) stabilized SeNPs inhibited Aβ aggregation and also disaggregated amyloid fibrils into nontoxic amorphous oligomers in vitro [117]. In rat hippocampal regions, solid lipid NPs loaded with chrysin were shown to protect against oxidative stress caused by protein Aβ fragments (residues 25-35). The work of Wang et al. [118] examined the effect of selenium-containing clioquinol derivatives on Cu2+-induced oxidation of Aβ as well as on the scavenging activity of hydrogen peroxide, ROS production in the intracellular environment, and the aggregation of Aβ in SH-SY5Y neuroblastoma cells.
In vivo Models for Alzheimer's Disease
Biomarkers and ultrastructural changes in AD have been characterized in various animal models to understand the fundamental mechanisms underlying AD and to bridge the gap between clinical outcomes and neuropathological diagnosis [119]. Atrophy of the brain is the central indicator of AD. In most cases, mice models consist of transgenic lines with familial mutations that cause the development of brain amyloidosis [119]. Several methods of detecting amyloid plaques include MRI, multiphoton imaging, near-infrared fluorescence imaging [120], and positron emission tomography imaging [121, 122].
While the above techniques have some disadvantages (lack of similarity in clinical human outcomes), they may help analyze the prognosis of animals with AD. Observation of Aβ accumulation in vivo remains the gold standard for in vivo evaluation of AD. There have also been reports of some alterations in the brains of transgenic animals, such as differences in water diffusion and decreased levels of N-acetylaspartate (NAA) [123, 124]. However, it is not certain that treatment options will not need such biomarkers to evaluate therapeutic outcomes.
It should be noted that animal models are only capable of reproducing certain clinical features of AD, such as the formation and accumulation of amyloid plaques, and not the overall disease process with the loss of neurons. It is for this reason that animal experiments cannot be translated into clinical trials [125]. It has been typical in animal studies of AD to use transgenic mice expressing the human APP and PS1 with mutations. Only a few studies have transgenic mice that express both tangles and plaques generated [126].
Additionally, APP-expression models show plaque formation in the frontal and temporal cortex, hippocampus, and cerebellum, synaptic impairment, congophilic amyloid angiopathy, and memory loss [127]. As APP is essential for the formation of amyloid plaques, the mouse strain and promoter play a significant role in determining the specific phenotype of the mouse. Several types of transgenic mice have been developed as models of AD (such as Tg2576176 [128] and APP23 [129], 5xFAD [130]), each of which has different mutations that lead to different pathophysiological outcomes.
Knock-in mice are being used for better in vivo AD modeling, and it depends on the humanization of Aβ and the knock-in of specific APP FAD mutations. Animal models offer a significant advantage over classical transgenic models as they avoid overexpression of APP. Nevertheless, it is essential to note that these animals exhibit pathological changes due to knocking in specific and multiple FAD mutations [131]. It is also possible to study AD pathology using transgenic rats, although their genetics, physiology, and morphology are much more similar to those of humans. They have more brain tissue, which makes imaging the changes and collecting samples more convenient. The transgenic rat models showing amyloid plaques and the TgF344-AD rats with neurofibrillary tangles indicate that human tau is very similar to rat tau [131].
There is a drawback to transgenic mouse models in that they can cause familial AD. It is estimated that only 1% of human AD patients possess this characteristic; thus, misinterpretations may occur in clinical trials due to this lack of characteristic [132]. A crucial histopathological hallmark of AD is neurofibrillary tangles, formed when hyperphosphorylated tau protein aggregates intraneuronal and is absent in most AD-transgenic models. By impairing synaptic plasticity, neurofibrillary tangles are the primary cause of cognitive impairment in the brain. Progressive neuronal loss in AD cannot be replicated in transgenic animals as it is in humans. Instead, these animal models provide valuable insights into the role of oxidative stress in disease, inflammation, and mitochondrial dysfunction [133]. Other test animals used in AD experiments include mouse lemurs, rhesus monkeys 193, and dogs. Different researchers have evaluated those experiments. However, neither model can be categorized as entirely suitable for AD translation [133].
Future Perspective on Alzheimer's Disease
The disease is characterized by the deposition of amyloid fibrils within extracellular plaques containing the A peptide. The use of small-molecule inhibitors for treating AD is believed to be beneficial. It is worth noting, however, that some inhibitors are intrinsically ineffective since they cannot pass through the BBB. NPs, which take advantage of the physiological mechanisms of transcytosis, such as receptors and adsorptive channels, assist in the transcellular delivery of small molecules, drugs, and markers. It has been recently discovered that iron-chelating compounds can penetrate the BBB with minimal neurotoxicity when incorporated into NPs [134]. As a result of their conjugation to chelators, these NPs were found to have an unusual ability to cross the BBB, chelate metal ions, and then exit through the BBB with their complex metal ions. An effective way to reduce metal loading in neural tissue, thus preventing oxidative damage and its consequences in AD brain tissue, may be discovered with this technique. The development of NPs would allow for increased accumulation and release of pharmacological agents at pathological sites, water solubility, an improvement in pharmacokinetics, stability under physiological conditions, and bioavailability, as well as a reduction in side effects by limiting their location in unhealthy tissues [135, 136]. Additionally, it is recommended that NP formulations be more efficient if they are designed to exploit their differential binding to monomers and oligomers to achieve a specific inhibition.
Conclusion
It is widely agreed that nanotechnology holds great promise for revolutionizing AD treatment, as it can address the limitations associated with the conventional delivery of drugs. By enhancing pharmacokinetics, nanoplatforms deliver drugs to the CNS with minimal toxicity while allowing controlled drug release. Nanotechnology therapeutic molecules can be delivered more efficiently and effectively to brain pathology sites by passing across the BBB. In AD therapeutics, nanotechnology heralds a new era of optimism and possibilities. Investing in research, collaborating across disciplines, and undergoing rigorous clinical evaluation remains essential to translating these promising advances into tangible benefits for people with AD. The progression of AD can indeed be prevented, retarded, or halted with concerted efforts, giving hope to millions of people worldwide.
Funding
No funding was received to conduct this study.
Authors Contributions
Conceptualization: Omranzadeh A; Methodology: all authors; Formal analysis and investigation: all authors; Writing – original draft preparation: all authors; Writing – review and editing: all authors; Funding acquisition: self-funding; Supervision: Omranzadeh A. All authors checked and approved the final version of the manuscript for publication in the present journal.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethical Approval
All applicable international, national, and/or institutional guidelines for writing a review paper were followed.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rahman A, Salam F, Islam MA, et al. Alzheimer s disease-an update. Bangladesh J Neurosci. 2013;28(1):52-8.
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015;11(3):332-84.
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018;14(3):367-429.
- Correction Naghavi M, Wang H, Lozano R, Davis A, Liang X, Zhou M, et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. The Lancet. 2015;385(9963):117-71.
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595-608.
- Shafiee M, Arekhi S, Omranzadeh A, et al. Saffron in the treatment of depression, anxiety and other mental disorders: Current evidence and potential mechanisms of action. J Affect Disord. 2018;227:330-337.
- Mehta M, Adem A, Sabbagh M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:728983.
- Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet. 2002;41(10):719-39.
- Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379-97.
- Arias JL. Nanotechnology and Drug Delivery: Nanoplatforms in Drug Delivery. 1st ed. Boca Raton: CRC Press; 2014. Chapter 1: Key Aspects in Nanotechnology and Drug Delivery; p. 1-27.
- Suri K, Wolfram J, Shen H, et al. Novel Approaches and Strategies for Biologics, Vaccines and Cancer Therapies. 1st ed. Massachusetts: Academic Press; 2015. Chapter 3: Advances in nanotechnology-based drug delivery platforms and novel drug delivery systems; p. 41-58.
- Singh J, Nandan PK. Drug Delivery System Using Nanotechnology for Alzheimer’s Disease. 2018:407.
- Parveen S, Sahoo SK. Nanomedicine: clinical applications of polyethylene glycol conjugated proteins and drugs. Clin Pharmacokinet. 2006;45(10):965-88.
- Orive G, Hernández RM, Rodríguez Gascón A, et al. Drug delivery in biotechnology: present and future. Curr Opin Biotechnol. 2003;14(6):659-64.
- Sadr S, Lotfalizadeh N, Abbasi AM, et al. Challenges and prospective of enhancing hydatid cyst chemotherapy by nanotechnology and the future of nanobiosensors for diagnosis. Trop Med Infect Dis. 2023;8(11):494.
- Sadr S, Poorjafari JP, Haratizadeh MJ, et al. Current status of nano-vaccinology in veterinary medicine science. Vet Med Sci. 2023;9(5):2294-308.
- Saeed M, Sadr S, Gharib A, et al. Phytosomes: A promising nanocarrier for enhanced delivery of herbal compounds in cancer therapy. J Lab Anim Res. 2022;1(1):26-32.
- Soleymani N, Sadr S, Santucciu C, et al. Evaluation of the In-Vitro Effects of Albendazole, Mebendazole, and Praziquantel Nanocapsules against Protoscolices of Hydatid Cyst. Pathogens. 2024;13(9):790.
- Sorouri N, Soleymani N, Sadr S, et al. Investigating the therapeutic effects of curcumin nanocapsules in hydatid cyst-infected mice. Exp Parasitol. 2024;267:108860.
- Safari J, Zarnegar Z. Advanced drug delivery systems: Nanotechnology of health design A review. J Saudi Chem Soc. 2014;18(2):85-99.
- Betancourt T, Doiron A, Homan KA, et al. Controlled release and nanotechnology. 10th ed. New York: Springer; 2009. Chapter: Nanotechnology in drug delivery; p. 283-312.
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741-66.
- Miyakawa T, Shimoji A, Kuramoto R, et al. The relationship between senile plaques and cerebral blood vessels in Alzheimer’s disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982;40(2):121-9.
- Kawai M, Kalaria RN, Harik SI, et al. The relationship of amyloid plaques to cerebral capillaries in Alzheimer’s disease. Am J Pathol. 1990;137(6):1435-46.
- Paroni G, Bisceglia P, Seripa D. Understanding the Amyloid Hypothesis in Alzheimer’s Disease. J Alzheimers Dis. 2019;68(2):493-510.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353-6.
- Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis. 2007;4(1):13-27.
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631-9.
- Bush AI. The metal theory of Alzheimer’s disease. J Alzheimers Dis. 2013;33 Suppl 1:S277-81.
- Gouras GK, Beal MF. Metal chelator decreases Alzheimer beta-amyloid plaques. Neuron. 2001;30(3):641-2.
- Huang X, Cuajungco MP, Atwood CS, et al. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274(52):37111-6.
- Lovell MA, Robertson JD, Teesdale WJ, et al. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158(1):47-52.
- Cherny RA, Legg JT, McLean CA, et al. Aqueous dissolution of Alzheimer’s disease Abeta amyloid deposits by biometal depletion. J Biol Chem. 1999;274(33):23223-8.
- Cherny RA, Atwood CS, Xilinas ME, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30(3):665-76.
- Bartus RT, Emerich DF. Cholinergic markers in Alzheimer disease. JAMA. 1999;282(23):2208-9.
- Rossor MN, Emson PC, Mountjoy CQ, et al. Reduced amounts of immunoreactive somatostatin in the temporal cortex in senile dementia of Alzheimer type. Neurosci Lett. 1980;20(3):373-7.
- Henke H, Lang W. Cholinergic enzymes in neocortex, hippocampus and basal forebrain of non-neurological and senile dementia of Alzheimer-type patients. Brain Res. 1983;267(2):281-91.
- Soininen H, Kosunen O, Helisalmi S, et al. A severe loss of choline acetyltransferase in the frontal cortex of Alzheimer patients carrying apolipoprotein epsilon 4 allele. Neurosci Lett. 1995;187(2):79-82.
- Gattaz WF, Forlenza OV, Talib LL, et al. Platelet phospholipase A(2) activity in Alzheimer’s disease and mild cognitive impairment. J Neural Transm (Vienna). 2004;111(5):591-601.
- Gattaz WF, Talib LL, Schaeffer EL, et al. Low platelet iPLA₂ activity predicts conversion from mild cognitive impairment to Alzheimer’s disease: a 4-year follow-up study. J Neural Transm (Vienna). 2014;121(2):193-200.
- Gattaz WF, Cairns NJ, Levy R, et al. Decreased phospholipase A2 activity in the brain and in platelets of patients with Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1996;246(3):129-31.
- Weingarten MD, Lockwood AH, Hwo SY, et al. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72(5):1858-62.
- Gong CX, Grundke-Iqbal I, Iqbal K. Targeting tau protein in Alzheimer’s disease. Drugs Aging. 2010;27(5):351-65.
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663-72.
- Johnstone M, Gearing AJ, Miller KM. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol. 1999;93(1-2):182-93.
- Rogers J, Lue LF. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem Int. 2001;39(5-6):333-40.
- Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging. 2001;22(6):903-8.
- McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21(2):195-218.
- Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol. 2003;27(3):325-55.
- Iqbal K, Alonso Adel C, Chen S, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739(2-3):198-210.
- Delbreil P, Rabanel JM, Banquy X, et al. Therapeutic nanotechnologies for Alzheimer’s disease: A critical analysis of recent trends and findings. Adv Drug Deliv Rev. 2022;187:114397.
- Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci. 2020;23(10):1183-1193.
- Sensi SL, Granzotto A, Siotto M, et al. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol Sci. 2018;39(12):1049-63.
- Medeiros R, Baglietto-Vargas D, LaFerla FM. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther. 2011;17(5):514-24.
- d’Errico P, Meyer-Luehmann M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front Aging Neurosci. 2020;12:265.
- Mahmood A, Miron VE. Microglia as therapeutic targets for central nervous system remyelination. Curr Opin Pharmacol. 2022;63:102188.
- Hickman S, Izzy S, Sen P, et al. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359-69.
- Füger P, Hefendehl JK, Veeraraghavalu K, et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci. 2017;20(10):1371-76.
- Wan J, Fu AK, Ip FC, et al. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: implications in Alzheimer’s disease. J Neurosci. 2010;30(20):6873-81.
- Chiba T, Yamada M, Aiso S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin Ther Targets. 2009;13(10):1155-67.
- Singh N, Das B, Zhou J, et al. Targeted BACE-1 inhibition in microglia enhances amyloid clearance and improved cognitive performance. Sci Adv. 2022;8(29):eabo3610.
- Stanciu GD, Luca A, Rusu RN, et al. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules. 2019;10(1):40.
- Vasic V, Barth K, Schmidt MHH. Neurodegeneration and Neuro-Regeneration-Alzheimer’s Disease and Stem Cell Therapy. Int J Mol Sci. 2019;20(17):4272.
- Kopeikina KJ, Carlson GA, Pitstick R, et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol. 2011;179(4):2071-82.
- Moreira PI, Carvalho C, Zhu X, et al. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta. 2010;1802(1):2-10.
- Szepesi Z, Manouchehrian O, Bachiller S, et al. Bidirectional Microglia-Neuron Communication in Health and Disease. Front Cell Neurosci. 2018;12:323.
- Yao J, Irwin RW, Zhao L, et al. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106(34):14670-5.
- Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2018;62(3):1403-16.
- Khayyatzadeh SS, Omranzadeh A, Miri-Moghaddam MM, et al. Dietary antioxidants and fibre intake and depressive symptoms in Iranian adolescent girls. Public Health Nutr. 2021;24(17):5650-56.
- Zabel M, Nackenoff A, Kirsch WM, et al. Markers of oxidative damage to lipids, nucleic acids and proteins and antioxidant enzymes activities in Alzheimer’s disease brain: A meta-analysis in human pathological specimens. Free Radic Biol Med. 2018;115:351-60.
- Omranzadeh A, Baradaran A, Ghodsi A, et al. Neutrophil-to-Lymphocyte Ratio as an Inflammatory Marker in Familial Mediterranean Fever: A Systematic Review and Meta-analysis. J Child Sci. 2021;11:e100-9.
- Ionescu-Tucker A, Cotman CW. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol Aging. 2021;107:86-95.
- Wang X, Wang W, Li L, et al. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240-7.
- Ferreira ME, de Vasconcelos AS, da Costa Vilhena T, et al. Oxidative Stress in Alzheimer’s Disease: Should We Keep Trying Antioxidant Therapies? Cell Mol Neurobiol. 2015;35(5):595-614.
- Moosmann B, Behl C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs. 2002;11(10):1407-35.
- Arya M, Kumar MKM, Sabitha M, et al. Nanotechnology approaches for enhanced CNS delivery in treating Alzheimer’s disease. J. Drug Deliv. Sci. Technol. 2019;51:297-309.
- Wen MM, El-Salamouni NS, El-Refaie WM, et al. Nanotechnology-based drug delivery systems for Alzheimer’s disease management: Technical, industrial, and clinical challenges. J Control Release. 2017;245:95-107.
- Zhao Z, Nelson AR, Betsholtz C, et al. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015;163(5):1064-1078.
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723-38.
- Sood S, Jain K, Gowthamarajan K. Intranasal therapeutic strategies for management of Alzheimer’s disease. J Drug Target. 2014;22(4):279-94.
- Mittal D, Ali A, Md S, et al. Insights into direct nose to brain delivery: current status and future perspective. Drug Deliv. 2014;21(2):75-86.
- Spuch C, Navarro C. The therapeutic potential of microencapsulate implants: patents and clinical trials. Recent Pat Endocr Metab Immune Drug Discov. 2010;4(1):59-68.
- Sigurdsson EM. Tau Immunotherapies for Alzheimer’s Disease and Related Tauopathies: Progress and Potential Pitfalls. J Alzheimers Dis. 2018;64(s1):S555-65.
- Lim SB, Banerjee A, Önyüksel H. Improvement of drug safety by the use of lipid-based nanocarriers. J Control Release. 2012;163(1):34-45.
- Zielińska A, Carreiró F, Oliveira AM, et al. Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules. 2020;25(16):3731.
- Carradori D, Balducci C, Re F, et al. Antibody-functionalized polymer nanoparticle leading to memory recovery in Alzheimer’s disease-like transgenic mouse model. Nanomedicine. 2018;14(2):609-18.
- Lane LA, Qian X, Nie S. SERS Nanoparticles in Medicine: From Label-Free Detection to Spectroscopic Tagging. Chem Rev. 2015;115(19):10489-529.
- Stuart MAC, Huck WT, Genzer J, et al. Emerging applications of stimuli-responsive polymer materials. Nature Mater. 2010;9:101-13.
- McClements DJ. Nanoemulsions versus microemulsions: terminology, differences, and similarities. Soft matter. 2012;8:1719-29.
- Kaur A, Nigam K, Srivastava S, et al. Memantine nanoemulsion: a new approach to treat Alzheimer’s disease. J Microencapsul. 2020;37(5):355-65.
- Chanphai P, Bekale L, Sanyakamdhorn S, et al. PAMAM dendrimers in drug delivery: loading efficacy and polymer morphology. Can J Chem. 2017;95(9):891-6.
- Kojima C, Turkbey B, Ogawa M, et al. Dendrimer-based MRI contrast agents: the effects of PEGylation on relaxivity and pharmacokinetics. Nanomedicine. 2011;7(6):1001-8.
- Shcharbin DG, Klajnert B, Bryszewska M. Dendrimers in gene transfection. Biochemistry (Mosc). 2009;74(10):1070-9.
- Yin T, Yang L, Liu Y, et al. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood-brain barrier permeability peptide-B6 peptide for potential use in Alzheimer’s disease. Acta Biomater. 2015;25:172-83.
- Balaban H, Nazıroğlu M, Demirci K, et al. The Protective Role of Selenium on Scopolamine-Induced Memory Impairment, Oxidative Stress, and Apoptosis in Aged Rats: The Involvement of TRPM2 and TRPV1 Channels. Mol Neurobiol. 2017;54(4):2852-68.
- Du H, Guo L, Yan S, et al. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670-5.
- Karakoti AS, Singh S, Kumar A, et al. PEGylated nanoceria as radical scavenger with tunable redox chemistry. J Am Chem Soc. 2009;131(40):14144-5.
- Celardo I, Pedersen JZ, Traversa E, et al. Pharmacological potential of cerium oxide nanoparticles. Nanoscale. 2011;3(4):1411-20.
- Chigurupati S, Mughal MR, Okun E, et al. Effects of cerium oxide nanoparticles on the growth of keratinocytes, fibroblasts and vascular endothelial cells in cutaneous wound healing. Biomaterials. 2013;34(9):2194-201.
- Kwon HJ, Cha MY, Kim D, et al. Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer’s Disease. ACS Nano. 2016;10(2):2860-70.
- Lowe TL, Strzelec A, Kiessling LL, et al. Structure-function relationships for inhibitors of beta-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry. 2001;40(26):7882-9.
- Hilbich C, Kisters-Woike B, Reed J, et al. Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease beta A4 peptides. J Mol Biol. 1992;228(2):460-73.
- Soto C, Kindy MS, Baumann M, et al. Inhibition of Alzheimer’s amyloidosis by peptides that prevent beta-sheet conformation. Biochem Biophys Res Commun. 1996;226(3):672-80.
- Plissonneau M, Pansieri J, Heinrich-Balard L, et al. Gd-nanoparticles functionalization with specific peptides for ß-amyloid plaques targeting. J Nanobiotechnology. 2016;14(1):60.
- Wengenack TM, Jack CR Jr, Garwood M, et al. MR microimaging of amyloid plaques in Alzheimer’s disease transgenic mice. Eur J Nucl Med Mol Imaging. 2008;35 Suppl 1:S82-8.
- Sancey L, Kotb S, Truillet C, et al. Long-term in vivo clearance of gadolinium-based AGuIX nanoparticles and their biocompatibility after systemic injection. ACS Nano. 2015;9(3):2477-88.
- Elias A, Tsourkas A. Imaging circulating cells and lymphoid tissues with iron oxide nanoparticles. Hematology Am Soc Hematol Educ Program. 2009:720-6.
- Singh N, Jenkins GJ, Asadi R, et al. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 2010;1(1).
- Kabanov AV, Gendelman HE. Nanomedicine in the diagnosis and therapy of neurodegenerative disorders. Prog Polym Sci. 2007;32(8-9):1054-82.
- Bastus NG, Kogan MJ, Amigo R, et al. Gold nanoparticles for selective and remote heating of β-amyloid protein aggregates. Mater Sci Eng C. 2007;27(5-8):1236-40.
- Kogan MJ, Bastus NG, Amigo R, et al. Nanoparticle-mediated local and remote manipulation of protein aggregation. Nano Lett. 2006;6(1):110-5.
- Liao YH, Chang YJ, Yoshiike Y, et al. Negatively charged gold nanoparticles inhibit Alzheimer’s amyloid-β fibrillization, induce fibril dissociation, and mitigate neurotoxicity. Small. 2012;8(23):3631-9.
- Neely A, Perry C, Varisli B, et al. Ultrasensitive and highly selective detection of Alzheimer’s disease biomarker using two-photon Rayleigh scattering properties of gold nanoparticle. ACS Nano. 2009;3(9):2834-40.
- Ramassamy C, Longpré F, Christen Y. Ginkgo biloba extract (EGb 761) in Alzheimer’s disease: is there any evidence? Curr Alzheimer Res. 2007;4(3):253-62.
- Ze Y, Hu R, Wang X, et al. Neurotoxicity and gene-expressed profile in brain-injured mice caused by exposure to titanium dioxide nanoparticles. J Biomed Mater Res A. 2014;102(2):470-8.
- Schweizer U, Bräuer AU, Köhrle J, et al. Selenium and brain function: a poorly recognized liaison. Brain Res Brain Res Rev. 2004;45(3):164-78.
- Zhang J, Zhou X, Yu Q, et al. Epigallocatechin-3-gallate (EGCG)-stabilized selenium nanoparticles coated with Tet-1 peptide to reduce amyloid-β aggregation and cytotoxicity. ACS Appl Mater Interfaces. 2014;6(11):8475-87.
- Wang Z, Wang Y, Li W, et al. Design, synthesis, and evaluation of multitarget-directed selenium-containing clioquinol derivatives for the treatment of Alzheimer’s disease. ACS Chem Neurosci. 2014;5(10):952-62.
- Delatour B, Epelbaum S, Petiet A, et al. In vivo imaging biomarkers in mouse models of Alzheimer’s disease: are we lost in translation or breaking through? Int J Alzheimers Dis. 2010 ;2010:604853.
- Hintersteiner M, Enz A, Frey P, et al. In vivo detection of amyloid-beta deposits by near-infrared imaging using an oxazine-derivative probe. Nat Biotechnol. 2005;23(5):577-83.
- Dong J, Revilla-Sanchez R, Moss S, et al. Multiphoton in vivo imaging of amyloid in animal models of Alzheimer’s disease. Neuropharmacology. 2010;59(4-5):268-75.
- Spires TL, Meyer-Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25(31):7278-87.
- Sun SW, Song SK, Harms MP, et al. Detection of age-dependent brain injury in a mouse model of brain amyloidosis associated with Alzheimer’s disease using magnetic resonance diffusion tensor imaging. Exp Neurol. 2005;191(1):77-85.
- Marjanska M, Curran GL, Wengenack TM, et al. Monitoring disease progression in transgenic mouse models of Alzheimer’s disease with proton magnetic resonance spectroscopy. Proc Natl Acad Sci U S A. 2005;102(33):11906-10.
- Banik A, Brown RE, Bamburg J, et al. Translation of Pre-Clinical Studies into Successful Clinical Trials for Alzheimer’s Disease: What are the Roadblocks and How Can They Be Overcome? J Alzheimers Dis. 2015;47(4):815-43.
- Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275(3):251-83.
- Drummond E, Wisniewski T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 2017;133(2):155-175.
- Elfenbein HA, Rosen RF, Stephens SL, et al. Cerebral beta-amyloid angiopathy in aged squirrel monkeys. Histol Histopathol. 2007;22(2):155-67.
- Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43-51.
- Heuer E, Rosen RF, Cintron A, et al. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr Pharm Des. 2012;18(8):1159-69.
- Di Marco LY, Farkas E, Martin C, et al. Is Vasomotion in Cerebral Arteries Impaired in Alzheimer’s Disease? J Alzheimers Dis. 2015;46(1):35-53.
- Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14(10):1097-105.
- Lee YJ, Han SB, Nam SY, et al. Inflammation and Alzheimer’s disease. Arch Pharm Res. 2010;33(10):1539-56.
- Yu YJ, Zhang Y, Kenrick M, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3(84):84ra44.
- Rizzo LY, Theek B, Storm G, et al. Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr Opin Biotechnol. 2013;24(6):1159-66.
- Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8(6):2101-41.
