Morshed S1*, Jahan S1, Hasan M2, Yadav A1, Faisal I1, Hasanat MA1 and Fariduddin M1
1Department of Endocrinology, Bangabandhu Sheikh Mujib Medical University (BSMMU), Bangladesh
2National Institute of Neuroscience (NINS), Bangladesh
*Correspondence: Shahed Morshed, Department of Endocrinology, Bangabandhu Sheikh Mujib Medical University (BSMMU), Bangladesh
Received on 18 June 2019; Accepted on 19 July 2019; Published on 24 July 2019
Copyright © 2019 Morshed S, et al. This is an open access article and is distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
We report a 26-year-old male who was initially diagnosed as osteopetrosis and referred for endocrine evaluations. But due to specific clinical features supported by investigations, we diagnosed the case as sclerosteosis. The patient was managed by a multidisciplinary team approach. Sclerosteosis may be erroneously diagnosed as osteopetrosis. However, many unique features make it a separate entity.
Keywords
sclerosteosis, osteopetrosis, Van Buchem disease, syndactyly, Truswell-Hansen disease, hyperostosis
Abbreviations
SOST: sclerostin; LRP4: low-density lipoprotein receptor-related protein 4; OMIM: online mendelian inheritance in man; LMN: lower motor neuron; ICP: intracranial pressure
Introduction
Sclerosteosis is one of the hereditary sclerosing bone disorders of intramembranous ossification often described as craniotubular hyperostosis-syndactyly syndrome [1]. Due to similarities of radiological features, it was first described as osteopetrosis with syndactyly and later named as sclerosteosis as a separate disease. It is often called as Truswell-Hansen disease that is derived from the names of the two clinicians who first described the disease [2]. Except few cases reported from different countries, all are from the Afrikaner community of South Africa and the Netherland due to their common ancestry [3]. The minimum prevalence is 1 in 60,000 in the Afrikaner community and gene frequency is 0.0035 with 10,000 clinically normal heterozygotes in this population [4, 5]. From a recent update, the number of reported cases are 96 [6].
Depending on mutation, two types of this disorder is described: type 1 (SOST-related, OMIM # 269500) and type 2 (LRP4-related, OMIM # 614305) sclerosteosis. The SOST gene is located in 17q12-q21 and the LRP4 gene is located in 11p11.2 respectively [7, 8]. As genetic analysis is not readily available in our setting, so we diagnosed the case as sclerosteosis by clinical, biochemical and radiological features.
Case Report
A 26-year-old Bangladeshi male, 3rd issue of non-consanguinous parents presented with progressively increasing headache, dimness of vision and hearing difficulty for 3½ years and feeling of sudden electric shock-like sensation in the left side of face triggered by touching lips and face for three months. He gave no history of fragility fracture, perioral paresthesia and carpopedal spasm, fatigue, recurrent infections and bleeding from any sites. All family members were clinically healthy. The patient could not remember any previous episodes of facial nerve palsy. The patient had midface hypoplasia with wide jaw (Figure 1), asymmetrical cutaneous syndactyly in both hands and feet, clinodactyly and nail dystrophy (Figure 2) which were present since birth. He also had hyposmia, bilateral optic atrophy (Figure 3), bilateral lower motor neuron (LMN) type 7th cranial nerve palsy and profound, mixed type of hearing loss with reduced compliance of tympanic membrane due to stiff middle ear ossicles (impedance audiometry) with left-sided trigeminal neuralgia. All routine investigations including liver enzymes and calcium panel were within normal limit except hypovitaminosis D.
Imaging of bones of different parts of the body revealed craniotubular hyperostosis (Figure 4, 5 and 6). Genetic analysis could not be done. The patient was referred to the department of Ophthalmology, Neurosurgery, Orthodontics, and Otolaryngology for further management. Features of trigeminal neuralgia improved with deflazacort and pregabalin. Exposed corneas were managed with artificial tear and night taping. The patient was advised for decompression of cranial nerves by the neurosurgeon, but he refused the treatment and was lost to follow up.
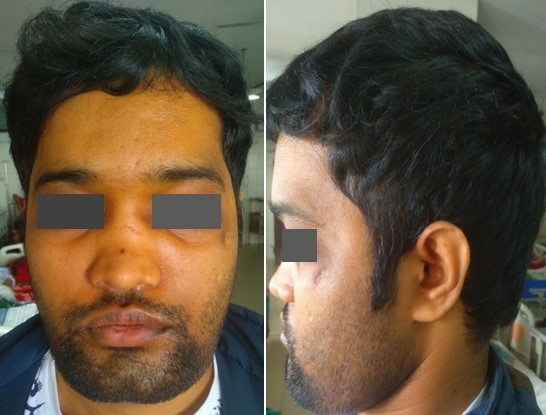
Figure 1: Square shaped jaw, hypertelorism and midface hypoplasia.
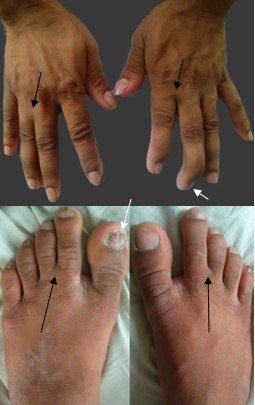
Figure 2: Syndactyly (black arrow) and nail hypoplasia (white arrow) are markers of sclerosteosis.
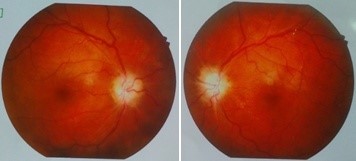
Figure 3: Bilateral optic atrophy due to entrapment neuropathy.
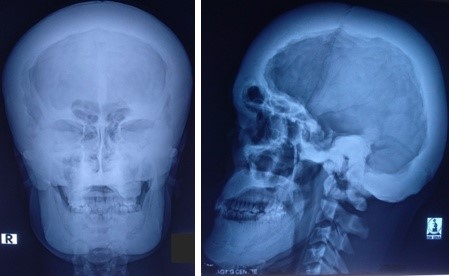
Figure 4: Marked cranial hyperostosis with endobone formation.
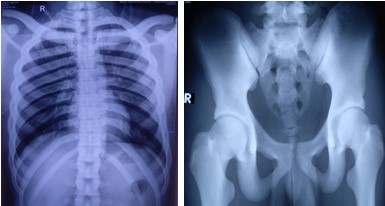
Figure 5: Diffuse sclerotic changes in all bones of pectoral and pelvic girdles.
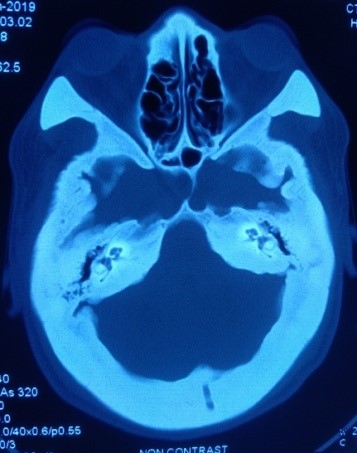
Figure 6: Cranial hyperostosis on CT scan of head.
Discussion
Sclerosteosis is an autosomal recessive disorder with progressive disfiguring skeletal and compressive neurological features from exaggerated bone formation [3]. The high bone mass is due to the absence of expression or secretion of sclerostin from mature osteocytes. Sclerostin acts as a potent antagonist of Wnt/β-catenin signaling after binding with Wnt coreceptor LRP5/6 and interaction with LRP4 [9].
The clinical and radiological features of sclerosteosis may be similar in different diseases. Due to different natural history and complications, it is essential to differentiate this disease from similar diseases. The main differential diagnoses with some differentiating traits are provided (Table 1). Presence of syndactyly and prominent jaw involvement separate this disorder from osteopetrosis (Albers-Schonberg disease). Syndactyly may be present in osteopathia striata with cranial sclerosis (Voorhoeve disease) but absence of extraskeletal features and striations along the long bone made this disorder unlikely in our patient.
Although the presenting features of this patient were consistent with sclerosteosis but there was no history of consanguinity of marriage between parents and absence of tall stature (patient’s height 171 cm). A case of sclerosteosis in a 45-year-old female described by Ahmed et al. [10] from Bangladesh had asymmetrical face with polydactyly in one hand, unilateral sensory type trigeminal and LMN type facial nerve palsy but without optic nerve atrophy. To our best knowledge, this was the first and only reported case from Bangladesh until date.
Sclerosteosis is associated with significant mortality and morbidity that were observed from the natural history of 63 persons for 38 years. It was found that syndactyly (76%) was not a consistent but characteristic feature of sclerosteosis [3]. As type 2 sclerosteosis is described in only 4 cases, the phenotypic differences with type 2 sclerosteosis are not well established. While syndactyly of 2nd and 3rd fingers is the hallmark of type 1, syndactyly involving 2nd to 4th fingers is predominantly found in type 2 sclerosteosis [1]. In our patient, cutaneous sclerodactylies were present between 2nd and 3rd finger of all except right hand where it was present between 3rd and 4th fingers. Trigeminal neuralgia is relatively a rare presentation of sclerosteosis that was present in this case. It is usually refractory to medical treatment [11], but in our case, the pain responded well to corticosteroid and pregabalin. Another fatal complication is sudden death, which is caused by the impaction of the medulla oblongata in the foramen magnum due to the progressive rise of intracranial pressure (ICP) by bony overgrowth [3]. Although, cranial imaging revealed endobone formation but there were no other clinical and MRI features of raised ICP in our case except headache (Figure 7).
There is no definitive therapy for the disease till now. In early adulthood, prophylactic craniectomy may be done. Surgical decompression of cranial nerves and mandibular reconstruction can be performed with variable results. An external hearing aid may be helpful. Syndactyly can be removed by cosmetic repair [5, 14]. Without prophylactic craniectomy, the average age of death was 33 years [3].
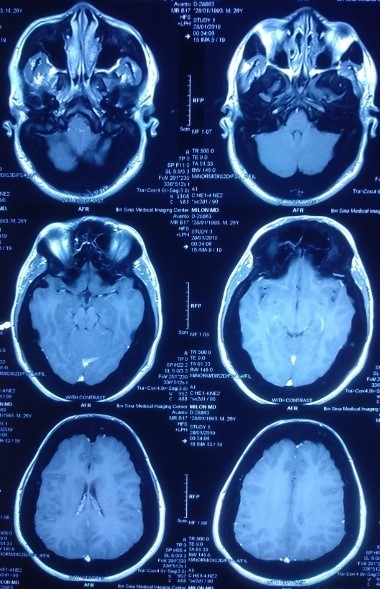
Figure 7: Normal MRI of brain.
| Similarity | Traits | Differential Diagnoses |
|---|
| Osteopetrosis [12] | Sclerosteosis |
|---|
| Radiological |
| Mandible | Less affected | Prominent feature |
| Syndactyly | Absent | Present |
| Pathological fracture | Common | Absent |
| Hematological Complications | Common | Absent |
| Hypocalcemia | Common | Absent |
| Nephrocalcinosis | May occur | Absent |
| | Van Buchem disease [13] | Sclerosteosis |
|---|
| Skeletal |
| Severity of bone changes | Mild | Mild to severe |
| Stature | Normal | Tall |
| Syndactyly | Absent | Present |
| | Osteopathia striata with cranial sclerosis [1] | Sclerosteosis |
|---|
| Syndactyly |
| Inheritance | X-linked recessive | Autosomal recessive |
| Complex digital malformation | Common | Rare |
| Extraskeletal anomalies | Present | Absent |
| Intellectual disability | Common | Absent |
| Striations along long axis of bone | Present | Absent |
Table 1: Differential diagnoses of sclerosteosis with key distinguishing features.
Conclusion
Early diagnosis of sclerosteosis is essential for the management of neurological complications and future genetic counseling. Due to unavailability of genetic analysis in our setting, characteristic clinical and radiological features along with the exclusion of other similar diseases are the mainstay of diagnosis.
Conflicts of Interest
The authors have no multiplicity of interest to disclose.
Funding Statement
There was no specific funding for this case report.
Acknowledgements
We are grateful to our patient for giving the consent to report the case.
References
- Yagi H, Takagi M, Hasegawa Y, et al. Sclerosteosis (craniotubular hyperostosis-syndactyly) with complex hyperphalangy of the index finger. Pediatr Radiol. 2015;45(8):1239-43.
- Amalnath SD, Vivekanandan M. Sclerosteosis (Truswell-Hansen disease). Indian J Hum Genet. 2013;19(2):270-72.
- Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet. 2003;63(3):192-97.
- Beighton P. Sclerosteosis. J Med Genet. 1988;25(3):200-03.
- Beighton P, Hamersma H. Sclerosteosis in South Africa. S Afr Med J. 1979;55(20):783-88.
- Appelman-Dijkstra N, Lierop AV, Papapoulos S. SOST-related sclerosing bone dysplasias. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2002 [Updated 2019 Mar 21].
- Balemans W, Ende JVD, Paes-Alves AF, et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet. 1999;64(6):1661-69.
- Leupin O, Piters E, Halleux C, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489-500.
- Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone. 2017;96:29-37.
- Ahmed S, Kader M, Ahmed Q, et al. A forty five years female patient with sclerosteosis. Journal of Medicine, 2010;11(2):207-11.
- de Andrade EM, Beer-Furlan A, Duarte KP, et al. Management of trigeminal neuralgia in sclerosteosis. Surg Neurol Int. 2013;4(Suppl 6):S455-S459.
- Wu CC, Econs MJ, DiMeglio LA, et al. Diagnosis and management of osteopetrosis: consensus guidelines from the osteopetrosis working group. J Clin Endocrinol Metab. 2017;102(9):3111-23.
- Jacobs P. Van Buchem disease. Postgrad Med J. 1977;53:506.
- du Plessis JJ. Sclerosteosis: neurosurgical experience with 14 cases. J Neurosurg. 1993;78(3):388-92.
