Abstract
Proton pump inhibitors (PPIs) are commonly prescribed in conjunction with other drugs as a preventative measure. Their primary function is acid suppression; as such, they subsequently hold back intestinal calcium absorption. By this mechanism, chronic use of PPIs is suggested to induce osteoporosis, a condition that gradually increases bone fragility and is associated with high suffering and morbidity rates. Additionally, PPI administration amongst elderly patients for more than one year has been shown to have an increased risk for hip fractures. This current review consists of literature searches from PubMed and the National Library of Medicine (USA). It analyzes factors such as age, race, gender, menopausal status, lifestyle, medical conditions, and polypharmacy, which may induce osteoporosis, and explores the mechanism of how PPIs may trigger osteoporosis development and/or progression. Clinicians should be cautious of severe osteoporosis as a potential consequence of PPI use, and be aware that it can be mitigated by the appropriate selection of pharmacological therapies and modalities.
Keywords
proton pump inhibitors, osteoporosis, acid-related disorders, fracture, osteoclast, osteoblast
Abbreviations
PPIs: proton pump inhibitors, GERD: gastroesophageal reflux disease, FDA: Food and Drug Administration, CKD: chronic kidney disease, BMD: bone mineral density, OC: osteoclast, OB: osteoblast, NSAIDs: nonsteroidal anti-inflammatory drugs, PTH: parathyroid hormone, M-CSF: macrophage (Mφ) colony-stimulating factor, TAM: tumor-associated macrophages, OPG: osteoprotegerin, LPA: lysophosphatidic acid, OCIF: osteoclastogenesis inhibitory factor, MMA: methylmalonic acid, PTHrP: parathyroid hormone-related peptide, PBMC: peripheral blood mononuclear cell
Introduction
Proton pump inhibitors (PPIs) are composed of a lipophilic base with pyridine and benzimidazole, having a pKa of 4-5, and principally function as acid suppressors. Omeprazole, the first PPI, was introduced in the late 1980s and is able to inhibit H+/K+ ATPase pump in the parietal cell and reduce gastric acid secretion [1]. New prodrug PPIs bind covalently with one or more cysteine residues of gastric H+/K+ ATPase in acidic conditions, converting cysteine into sulfuric acid or sulfonamides. Several PPIs are similar in structure (e.g., esomeprazole, pantoprazole, lansoprazole, rabeprazole, ilaprazole, and dexlansoprazole) but are different in pharmacodynamic profile, allowing them to be specific for various diseases [2]. They have been traditionally used as treatment for acid-related disorders, metabolic acidosis in the gastrointestinal tract, such as peptic ulcer disease, gastroesophageal reflux disease (GERD), duodenal ulcers, and erosive esophagitis. The United States Food and Drug Administration (FDA) also approved this drug to be used in Helicobacter pylori eradication and pathological hypersecretory conditions such as Zollinger–Ellison syndrome. PPIs are one of the most frequently used classes of medications in the world, and with their wide range of therapeutic indications, PPIs have seen a remarkable rise in use in the last few years.
While short-term administration of PPIs may lead to mild nausea, headache, diarrhea, abdominal cramp, constipation, flatulence, rash, and dizziness [3], prolonged administration of PPI has been reported to have a myriad of side effects, such as enteric infection, hypomagnesemia, B12 deficiency, iron deficiency, calcium deficiency, pneumonia, gastric carcinoids, gastric polyps, ischemic heart diseases, dementia, prominent acute interstitial nephritis, or chronic kidney disease (CKD) [4–7]. Development of CKD is shown to be dose-dependent, with patients taking two PPI doses per day having a higher risk of CKD than those taking a single PPI dose [4].
There has also been recent and growing attention on the postulation of PPI induction of osteoporosis, a bone disorder characterized by decreased bone mineral density (BMD), diminished bone mass, microarchitectural deterioration, and skeletal fragility. Some studies, such as Targownik et al. [8] and Kaye et al. [9], have stipulated that there is a lack of evidence to prove that osteoporosis or hip fracture results from years-long PPI administration.
However, it is notable that gastric acid plays a key role in the absorption of calcium. Since PPIs block gastric acid secretion and calcium is not solubilized or maximally absorbed at non-acidic pH, PPIs therefore effectively and directly hold back intestinal calcium absorption [10]. A study reveals a significant decrease in absorption of calcium carbonate in women with PPIs [10]. This impaired gastrointestinal calcium absorption predisposes PPI-using patients to curtailed bone strength and increased vulnerability to fractures [11]. PPI-induced gastrin production and increased hypochlorhydria can lead to bone remodeling, mineral absorption, and muscle strength. Vangala et al. [12] conducted a study of 4551 cases and 45,510 controls, and suggest a modestly increased risk of hip fracture among hemodialysis patients receiving PPIs (adjusted odds ratio, 1.19; 95% confidence interval, 1.11 to 1.28). Yang et al. [13] demonstrated that chronic PPI therapy for more than one year can increase the risk of bone fractures (particularly the hip in patients older than 65 years) and osteoporosis (by means of decreasing BMD). Yang et al. [13] is further supported by Corley et al. [14], who showed that chronic PPI therapy for more than 2 years can also induce hip fractures. Additionally, both the recent systematic reviews published by Thong et al. [15] and Hussain et al. [16] have demonstrated that there is a statistically significant positive association between use of PPIs and elevated fracture risk (RR 1.26 [95% CI, 1.17-1.35], p < 0.00001). In fact, an individual on PPIs was reported to have a 26% increased risk of hip fracture as compared to an individual who did not use PPIs [16]. Even nonsteroidal anti-inflammatory drugs (NSAIDs) users prescribed with these prophylactic agents may remain at risk of osteoporosis [17]. Additionally, dose-dependent therapy makes the old age group more susceptible to bone loss than the younger one [18].
Given the paucity of population-based epidemiological studies that focus on the association between high-dose PPIs and risk of fracture and/or osteoporosis, a direct relation between the two is still an enigma [19, 20]. A few in vitro animal studies and some human data suggest that PPIs decrease the osteoclast (OC) bone resorption activity as well as alter the bone-forming activity of osteoblast (OB) [18, 21]. In contrast, in vivo studies regarding the influence of PPI on bone metabolism are still less conclusive [22]. Despite these inconclusive results, the literature still marks PPI as influential on bone metabolism and may result in osteoporosis.
Thus, this study aims to determine the cellular effects of PPIs on osteoclast and osteoblast activity in the presence or absence of other important biological molecules or other drugs given to the patient due to comorbidity. It seeks to analyze factors such as molecular components, age, race, gender, menopausal status, lifestyle, medical conditions, and polypharmacy, and their relation and interspersed effects on the triggering of osteoporosis development and/or progression.
Bones are made of two layers: the external cortical bone and internal spongy trabecular bone. The main components of bone matrices are collagens. The natural bone matrix has an organic part and an inorganic part called hydroxyapatite and is predominantly composed of calcium phosphate.
Bone may appear inert, but it is actually dynamic in nature [23] and is continuously remodeling. Trabecular bones and compact bones are replaced within 3–4 years and 10 years, respectively [24]. Bones are lined by cells known as osteocytes and possess regulatory cells known as osteoblasts and osteoclasts. An osteoblast (OB) is responsible for bone remodeling by using minerals like calcium and phosphates. An osteoclast (OC) is a multinucleated cell that maintains the serum calcium and phosphate levels by breaking down or reabsorbing old bone [25].
Bone remodeling depends on serum calcium levels, which are maintained by a balance between parathyroid hormone (PTH), calcitonin, vitamin D, and vitamin B complex. PTH is produced in response to low serum calcium levels, and it increases bone resorption to release calcium into the bloodstream. On the other hand, calcitonin is produced by the thyroid gland in response to high serum calcium, which opposes the action of PTH and promotes bone formation. Finally, vitamin D promotes calcium absorption in the gut, so it increases serum calcium to promote bone formation and decreases bone resorption. People have their strongest, most dense bones, called peak bone mass, in their thirties [26]. Estrogens and androgens are believed to inhibit bone resorption, and low estrogen levels after menopause and low serum calcium may accelerate bone loss. Estrogen keeps the number and activity of osteoblasts higher than osteoclasts, so that more bone is made than removed. Alcohol consumption, smoking, and drugs like glucocorticoids decrease calcium absorption from the gut through antagonism of vitamin D and drugs like heparin and L-thyroxine. The regulation of bone modelling and remodeling occurs due to the interaction of various kinds of molecules like receptors-ligand, cell-cell interaction, gene-protein interaction, microRNA/exosomes, ion pumps, specifically ATPase pump, and a few hormones [27].
It is important that this entire mechanism of bone formation and resorption is balanced. Turbulent breakdown of bone in comparison to the formation of new bone results in porous bone with low bone density, known as osteoporosis. It is also notable that while there are several molecular factors in this mechanism that may be affected by PPIs, such molecular factors may vary person to person, leading to a large number of interindividual pharmacokinetic variations and manifestations of various effects. For instance, the CYP2C19 polymorphism can affect an individual’s metabolism of PPIs, and CYP2C19-mediated metabolism can produce marked interpatient variability in acid suppression, in drug-interaction potential, and in its clinical efficacy [28].
2.1 Osteoblast and osteoclast genesis
2.1.1 Reverse signaling between osteoblast and osteoclast
Two vital cells, the osteoblast and the osteoclast, are involved in the regulation of bone modelling and remodeling. The regulative relation of these cells is vice versa; they both control the production, formation, and maturation of each other. Molecules that mediate osteoclast-osteoblast interactions by simultaneous signal transduction in both cell types have not yet been identified. Bone cells and progenitor cells express some membrane receptors and ligands on their surface to maintain their activities. The transmembrane ligand ephrin 2 on OCs interacts with receptor tyrosine kinase ephrin B4 (ephB4) present on OBs and osteocytes. The inhibition of OC promoted by the expression of ephB4 on osteoblasts allows the OB differentiation, consequently blocking bone resorption [29, 30]. The signaling mediated by EPH (OC-OB) stimulates bone formation via OB differentiation and inhibits OC activation via reverse signaling through ephrinB2 into osteoclasts by obstructing the induction of Fos and its transcriptional target NFAT, and thereby inhibits bone resorption. Since Ephb2 (encoding ephrinB2) is identified as an NFAT target gene during osteoclast differentiation. NFAT is also able to induce receptor activator nuclear factor κβ ligand (RANKL). Osteogenesis is promoted by EphB4 signaling by some regulatory factors like Dlx5, Osx, and Runx2 [30].
2.2 Role of immune cells in bone formation
Immunogenic molecules such as the macrophage (Mφ) colony-stimulating factor (M-CSF) and monocyte attractant protein (MCP1or CCL2) secreted by OB play a role in OC genesis [31, 32]. These molecules help in differentiation, activation, migration, survival, etc. Both induce differentiation of OC’s from bone marrow precursors after binding to C-Fms receptors expressed on macrophages, specifically M-CSF. Additionally, M-CSF promotes expression of receptor activator of nucleofactor κβ (RANK) in bone marrow precursor. However, MCP1 expression is upregulated during the inflammatory action of bone and induces the proliferation of bone marrow precursors [33].
A study presents evidence that pantoprazole-dependent augmentation of myelopoiesis in hosts having tumors is dependent on an enhanced expression of M-CSF and receptors for M-CSF & GM-CSF in BMC [34]. Pantoprazole activates tumor-associated macrophages (TAM) to a tumoricidal state in a murine model [34]. Therefore, PPI such as pantoprazole is capable of altering M-CSF expression in order to immunopotentiation, which may induce differentiation of bone marrow into OC precursors, which may in turn increase bone resorption compared to bone formation, leading to increased risk of osteoporosis.
The RANKL is the membrane-bound protein of the OB. The RANKL protein binds with RANK after either proteolytic cleavage or alternative splicing. The regulatory factors of RANKL are cytokines, hormones, and growth factors, including PTH, estrogen, and inflammatory cytokines. On the other hand, RANK is the TNF family membrane protein expressed on OC progenitor cells, mature OCs, dendritic cells, and mammary gland [35]. The interaction of RANK/RANKL induces OCs differentiation, activation via JNK/AP1, JAK/NF κβ, c-myc, and calcineurin/NFATC1, src, and MKK6/p38/MITF signaling pathway [36]. Osteoprotegerin (OPG), a soluble decoy receptor for receptor activator of nuclear factor-kappaB ligand (RANKL), an osteoclast differentiation factor, inhibits both differentiation and function of osteoclasts [37]. RANKL are able to bind with leucine rich repeats containing G protein coupled receptor 4 (LGR4 or GPR4), which inhibits the proper interaction between RANK/RANKL and blocks the OC genesis. RANKL is also expressed on the surface of an activated cluster of differentiation (CD) 4+ T helper 1 (Th1) cells by involving a reverse signaling process. This interaction suppresses interferon γ secretion from Th1 cells. The MAPK p38 signaling also plays a crucial role in this pathway.
2.3 Role of lysophosphatidic acid in bone formation
Lysophosphatidic acid (LPA) is a potent activator of OC formation [38]. LPA is produced by various cells, such as platelets and OB, which help in the fusion of progenitor OC cells. OC genesis additionally reduces the apoptosis of OC. On the other hand, the apoptosis of OC induced by OB via the Fas-mediated extrinsic apoptotic pathway, which is upregulated by estrogen or female hormone [39]. It is observed that PPI usage is harmful in the general population and worse for those on no antiplatelet agent. This suggests that PPIs promote certain risk via an unknown mechanism that does not directly involve platelet aggregation. The underlying mechanism may probably involve dysregulation of vascular nitric oxide synthase [40]. Studies document the possible interaction of clopidogrel and PPIs, leading to a decrease in the antiplatelet efficacy of clopidogrel. Clopidogrel is a prodrug that requires cytochrome P450 2C19 (CYP 2C19) enzyme for its conversion to an active thiol metabolite. PPIs inhibit enzyme CYP 2C19, interfering with the conversion of clopidogrel into its active metabolite [41]. The CYP2C19 polymorphism affects the metabolism of PPIs, causing large interindividual pharmacokinetic variations. Differences in CYP2C19-mediated metabolism can produce marked interpatient variability in acid suppression, drug-interaction potential, and in its clinical efficacy [28]. Ultimately increasing the risk of platelet aggregation and activated platelet [42], which is in turn a source of LPA (v). Now, LPA leads to increased OC activity and bone resorption. LPA from activated platelets may enhance both tumor growth and cytokine-mediated bone destruction [43].
D2 isoform of vascular (H+) ATPase V0 domain (Atp6V0d2) is highly expressed in osteoclasts. Bone resorption relies on the extracellular acidification function of vacuolar (V-) ATPase proton pump(s) present in the plasma membrane of osteoclasts. Atp6V0d2 (d2), an isoform of the d subunit in the V-ATPase, showed 5-fold higher expression than that of Atp6V0d2 (d1) in mature osteoclasts, indicating a potential function in osteoclastic bone resorption [44]. It is noticed that this Atp6V0d2 mainly helps OC to mature. Atp6V0d2-/- mice are not able to produce cell fusion, whereas the cell differentiation remains the same. As a consequence, the multinucleated mature OCs remarkably reduce; however, the differentiation of OC or OC related gene expression remains unchanged in Atp6V0d2-/- mice [45]. Although additional experiments are needed to elucidate the molecular mechanisms by which d2 functions in osteoclasts. Findings from a study concluded d2 as a potential therapeutic target for the treatment of diseases caused by excessive activity of osteoclasts, such as osteoporosis and rheumatoid arthritis [46–48]. PPI may inhibit Atp6V0d2, creating a scenario similar to Atp6V0d2-/- mice, which reduces mature OCs, thus inhibiting bone resorption, which looks favorable for bone formation; however, it doesn’t affect OC differentiation.
PPI inhibited phenotype-related gene expression and functional parameters. For both cell types and their cellular function, i.e., osteoclastic resorption and the formation of mineralized deposits by osteoblastic cells, were more affected than proliferation-related parameters. The three PPIs showed similar qualitative and quantitative effects but displayed some differences in underlying intracellular signaling pathways. These results suggest that PPIs might have a direct deleterious effect on bone cells, with the possibility of decreased bone turnover [18]. Several mouse models with genetic deletion or mutation of V-ATPase proton pump subunits (e.g., ATP6i/ATP6V0a3, ATP6V0a1) display osteopetrosis due to dysfunctional osteoclasts. ATP6V0d2 knockout mice also display osteopetrosis [45]. A study demonstrated that Atp6v0d2 (d2) is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption, and that d2 directly interacts with Atp6i, while Atp6v0d2 deficiency affects differentiation and the v-ATPase activity of osteoclasts [44]. An essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. Bone modelling and remodeling specifically depend on prime homeostasis of intracellular calcium and phosphate, which are expressed in the plasma of all three cell types, and help in the cellular transportation system. The bone matrix fate is specifically determined by some osteotropic agents, mechanical stimulation, and intracellular pH, which controls the activity, expression, regulation, and cell surface abundance of plasma membrane transport systems [49].
Influence of PPI on Osteoporosis: In the Presence and Absence of Estrogen
Hormones have an important role in bone formation and resorption. Hormones such as PTH, calcitonin, and sex hormones (mainly estrogen) play a mechanistic role in bone modelling, bone remodeling, and calcium absorption. From previous content, it is clear that osteoporosis is a disorder more common amongst those of an older age. In each individual after the age of 30, bone resorption exceeds bone formation, and bone mass begins to diminish. This is a more common predicament amongst women following the onset of menopause than their male counterparts, as post-menopausal women in particular are more susceptible due to the diminishment of estrogen.
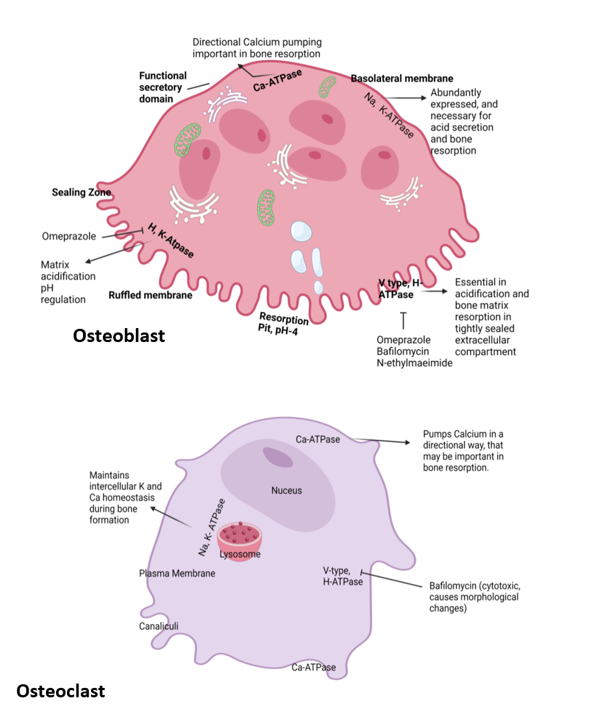 Figure 1: Location and function of ATPase in osteoclast and osteoblast and their inhibitors.
Figure 1: Location and function of ATPase in osteoclast and osteoblast and their inhibitors.
Role of Estrogen in Osteoporosis
Osteoclasts (Figure 1) and their precursor molecules are regulated by different signaling molecules. One of the key regulatory molecules is a RANK ligand, which is a membrane protein produced by a variety of cell types, including OBs and their precursor cells, as well as CD4+Th1 cells. RANK induces signaling pathways involved in the activation, differentiation, and survival of precursor cells for the formation of multinucleated OCs, as mentioned earlier in this study. The interaction between RANK ligand and RANK receptor promotes OC activity and survival, leading to increased resorption of bone. To balance this mechanism, the protein OPG is secreted from OB [50]. OPG secreted from OB is a cytokine receptor of the TNF family and is also an osteoclastogenesis inhibitory factor (OCIF). This OPG interacts with its specific ligand OPGL and blocks OC production [51]. The precursor cell of OPG is a B cell found in bone marrow. The expression of OPG is mainly supervised by cytokines, growth factors, and hormones like estrogen, vitamin D, and TNF. OPG acts deeply and binds with RANKL to decrease the interaction with the RANK and diminish the bone resorption. Various hormones and cytokines control the concentration of RANKL and OPG. PTH stimulates the production of RANKL and represses the expression of OPG, an effect thought to increase the activity of OCs [52]. High level of serum calcium activates calcitonin, which opposes the function of the PTH hormone in general [53], but after menopause, the action of estrogen reduces dramatically, and bone resorption increases, leading to bone fractures with osteoporosis [54]. Conversely, estrogen can increase the expression of OPG and inhibit RANKL signaling. An increase in bone resorption observed in states of estrogen deficiency in mice is mainly caused by a lack of ERα-mediated suppression of RANKL expression in bone lining cells [55]. These changes, which are thought to reduce the activity in OC’s, may contribute to estrogen’s ability to preserve or increase skeletal mass.
Omeprazole (30 mg/kg) was orally administered for eight weeks in ovariectomized rats, and the 16-week-old rats were then sacrificed. A low-calcium diet and PPI alter osteoclast activity and bone resorption signaling. The relative ratio of RANKL/OPG was increased in the group fed with a low-calcium and PPI (omeprazole-30 mg/kg body weight) diet. The bone formation marker osteocalcin was decreased, and the bone resorption marker, CTX-1 levels in serum [56]. Thus, PPI increases bone resorption chances in low estrogen conditions, which is often the case in post-menopausal women and almost all males. The total sex hormonal regulation is very important in osteoporosis induction in females, where a sudden reduction in a crucial molecule increases the susceptibility. That’s why osteoporosis is common in post-menopausal women. Although earlier studies showed that PPI use increases the risk of osteoporosis and bone fractures, another study shows the inhibitory effect of pantoprazole (a PPI variant) on OC formation and bone resorption both in vitro and in vivo. This suggests a potential therapeutic use of PPZ in the treatment of osteolytic disease with localized bone destruction [57]. Though this study does not include other factors such as hormones, cytokines, and M-CSF, which do change the impact of PPI on bone formation and resorption signaling.
Mechanism and Biochemistry Behind PPI-Inducing Osteoporosis
PPIs have pKa1 between 3.8 and 4.9 and are considered weak bases. This pKa enables PPI to get stored in the acidic space of the stimulated parietal cell, specifically in the secretory canaliculus, having a pH of 1.0. The accumulated PPI prodrug undergoes acid-dependent conversion into an activated reactive thiophilic reagent. Then it undergoes second protonation for its activation to the compounds that are capable of forming disulfides with accessible cysteines of the H, K-ATPase. The actual inhibitory form of PPI is a tetracyclic sulfenamide or sulfenic acid. The order of acid stability is tenatoprazole > pantoprazole > omeprazole > lansoprazole > rabeprazole (Table 1) [58].
| Drug | Cysteine |
| Omeprazole | cysteine 813 and cysteine 892 |
| Lansoprazole | cysteine 813 and cysteine 321 |
| Pantoprazole and tenatoprazole | cysteine 813 and cysteine 822 |
Table 1: Different PPIs are capable of binding to different cysteines of the H, K-ATPase.
Differences in PPI binding sites may modify its biological activity. PPIs show their activity on membrane transporters. They can induce osteoporosis by acting on targeted membrane transporters, present on osteoclast and osteoblast cells. A few H+ /K+ /ATPase pumps are present at the ruffled border membrane of OCs, which help to maintain OC activity. Other channels, such as K+ /Na+ gradients, Na+ channels, chloride bicarbonate exchange, etc., are also present. The acid environment of OC reduces the lucuma resorption and promotes bone mineralization [59]. The H+ /K+ /ATPase pump is involved in bone resorption. Omeprazole is able to suppress only ATP-dependent pumps 1000 times more in response than needed [60].
Another proton pump vascular H+ ATPase is present at the apical distribution of the ruffled membrane of avian and rat OCs [61]. In OCs, mRNA of V-type H+ ATPase is found in larger numbers and works simultaneously with the bicarbonate Cl channel [62]. Evidence shows that mutations in the gene encoding the a3 subunit of the proton pump cause infantile osteopetrosis [63]. Though PPIs are not a target-specific drug, they randomly block the H+ ion secretion and transportation via different mechanisms, which we have already described. By increasing pH, osteoclastic cells lose their working potential, and various biomolecules are activated due to high pH, which introduces osteoporosis in patients with prolonged PPI administration.
Effect of PPI on Vitamins and Minerals
8.1 PPIs and effects on vitamin B12 (VB12) absorption
Vitamin B12 is a dietary essential nutrient that we get from food supplements. PPI administration reduces absorption of essential nutrients, which may result in life-threatening diseases over time. PPIs majorly act on gastric secretion, which is essential for nutrient absorption. Vitamin B12 needs gastric acid to convert itself into an absorbing form [64, 65]. A short-term study revealed that B12 absorption reduces due to the presence of different acid suppressants [66]. There are several studies that have demonstrated that B12 absorption significantly decreases with chronic PPI exposure. VB12 has an on-induction osteoporosis. It helps to proliferate OB and their precursor cells and ALP, whereas a deficiency of B12 induces OC’s maturation and formation, helping in osteoporosis induction [67]. The in vitro experimental study shows that VB12 has no direct effect on OB, OC, as well as cellular differentiation, formation, or resorption activity in mouse bone marrow. Though elevated amounts of homocysteine and methylmalonic acid (MMA), which may be a result of B12 deficiency, induce osteoclast genesis as well as osteoporosis by increasing bone resorption [68]. The high level of homocysteine blocks some enzymes like lysyl oxidase, which plays a crucial role in collagen formation, especially in crosslinking in collagen fibres, by inactivating lysyl oxidase, B12 increases the risk of bone fracture and reduces BMD.
8.2 PPIs and effects on iron absorption
Prolonged PPI treatment can induce iron deficiency, which promotes osteoporosis via hypothyroidism [69]. Contradictory studies revealed that, in some cases, prolonged PPIs demonstration can induce anemia due to iron deficiency. A low iron diet, along with PPI-consuming individuals and rats, causes iron deficiency problems, which increase the PTH secretion [70]. Mutations in PTH are associated with severe abnormalities in chondrocyte proliferation [71]. Osteoporosis markedly delays the fracture healing process, mostly due to increased chondrocyte proliferation without a change in chondrocyte apoptosis [72]. Gastrin, increased by PPI, regulates parathyroid hormone-related peptide (PTHrP) [73], since PTHrP has a crucial involvement in bone metabolism and chondrogenesis [71, 74]. PPI may regulate bone formation and resorption via PTHrp via iron regulation.
8.3 PPIs and effects on calcium absorption/metabolism
Calcium is one of the key constructive units of bone. Maintaining a standard equilibrium between calcium and vitamin D improves the osteoporotic problem, as proven by long-term research [75]. Low calcium absorption may be the result of PPI administration. This is a more contradictory study than the B12 absorption. By lowering the absorption of calcium, the main constitutive element of bone, PPI disrupts bone microstructure, which is also an effect of hypochlorhydria (lowering the acid level of the stomach) [76]. PPI increases gastric acid pH, eventually causing hypochlorhydria that reduces calcium ionization as well as intestinal absorption. The calcium homeostasis regulates PTH, which is released from the parathyroid gland after sensing low concentrations of circulating calcium in the blood [26]. The calcium deficiency, along with vitamin D, induces secondary hyperparathyroidism. The vitamin D’s active hormonal form, namely calcitriol (1, 25 dihydroxy vitamin D), increases intestinal calcium as well as phosphorus absorption and inhibits the synthesis of PTH. There are dual pathways that induce hyperparathyroidism. Osteoporosis is one of the age-related problems. PPIs give a positive induction in the formation of osteoporosis by increasing gastrin and hypochlorhydria. Vitamin D deficiency occurs with aging and promotes bone disease along with PPIs administration before age 60 [77]. PTH elevates bone resorption by stimulating bone turnover markers like osteocalcin, alkaline phosphatase, tartrate-resistant acid phosphate, etc. Additionally, hydroxyproline diminishes calcium absorption and destroys bone microstructure via collagen structure distortion. The intestinal calcium absorption occurs via two mechanisms: active transport of calcium at the proximal duodenum induced by vitamin D and passive transport of calcium throughout the intestine due to the paracellular diffusion down a chemical gradient [78]. So, in brief, PPI therapy induces bone resorption in two ways: one via primary hyperparathyroidism through hypergastrinemia and parathyroid hyperplasia, another via secondary hyperparathyroidism, characterized by low calcium absorption, disrupted calcium balance in circulation, lowering the plasma [Ca++], etc. PTH overexpression is the result of primary and secondary hyperparathyroidism, which reduces bone strength and subsequently induces osteoporosis.
Effects of PPIs on Gene Expression and Enzymes
According to some studies, PPIs have an effective role in OBs and OCs genesis and function [18, 79]. A study concludes that a 1–10 μg/ml pantoprazole dose increases OB viability along with alkaline phosphatase activity, and the administration of 1–4 μg/ml omeprazole induces OB differentiation via increasing the level of osteocalcin. This study was carried out in both osteoblastic cells and MC3T3-E1 cells, both of which have given nearly equal results [21, 79]. PPIs also have a deleterious effect on osteoclast like osteoblast cells. It reduces expression of genes like c-myc, c-src, TRAP, and cathepsin K in peripheral blood mononuclear cell (PBMC) culture with 10-5M 10-3M omeprazole treatment for 21 days [18]. These remarked genes are especially expressed in osteoclasts. The 0.1–4 μg/ml omeprazole-treated osteoclast culture cells experiment shows suppression of calcitonin receptors, C-fos, nuclear factors of activated T cell (NFATc1), and matrix metalloproteinase 9 (MMP9) transcription as well as promotes the ratio of OPG to NFκβL in OB-treated culture media (MC3T3-E1) [21].
Summary and Conclusion
There are several PPIs such as esomeprazole, pantoprazole, lansoprazole, rabeprazole, ilaprazole, and dexlansoprazole with different pharmacodynamic profiles, viz, disease-specific [2]. Suppression of acid is the major function of PPI, which reduces intestinal calcium absorption and increases the risk of bone fragility [11]. PPI or H2RA administration over 2 years can induce hip fractures [14]. An association between PPI use and hip fracture risk has been described in a study conducted in 4551 cases and 45,510 controls [12]. At non-acidic pH, calcium does not solubilize or get absorbed, but rather remains in the solution [10]. Radiotracer study shows reduced calcium carbonate absorption in women due to PPI administration. Both animal and human data suggest that PPIs reduce the osteoclast (OC) bone resorption activity as well as change the bone-forming activity of osteoblast (OB) [18, 21]. Thus, the influence of PPI on total bone turnover needs to be focused on in the future.
PTH is produced due to low serum calcium levels, and it increases bone resorption to release calcium into the bloodstream. Calcitonin is produced by the thyroid gland in response to high serum calcium, which opposes the action of PTH and promotes bone formation. These molecular factors vary from person to person, which may cause variable interindividual effects of PPI between them [27]. Differences in the CYP2C19-mediated metabolism of PPI can produce marked interpatient variability in acid suppression, drug-interaction potential, and its clinical efficacy [28]. Osteoblasts and osteoclasts are involved in the regulation of bone modelling and remodeling, and they control each other’s proliferation. The signaling mediated by EPH (OC-OB) stimulates bone formation via OB differentiation and inhibits the OC activation via reverse signaling through ephrinB2 in osteoclasts. It obstructs Fos induction and its transcriptional target NFAT, inhibiting bone resorption. EphB4 signaling involves regulatory factors like Dlx5, Osx, and Runx2 [30].
Immunogenic molecules such as the M-CSF and monocyte attractant protein (MCP1or CCL2) secreted by OB play a role in OC genesis [31, 32]. M-CSF also promotes expression of receptor activator of nucleofector κβ (RANK) in bone marrow precursors. Pantoprazole activates TAM and alters M-CSF expression [34]. If the same occurs in bone marrow due to PPI, it may induce differentiation of bone marrow into OC’s via macrophage induction, leading to increased bone resorption compared to bone formation. Possible interaction of clopidogrel and PPIs leading to a decrease in the antiplatelet efficacy of clopidogrel [41]. Ultimately increases the risk of platelet aggregation and activated platelet, in turn, is a source of LPA (v) [42]. LPA leads to increased OC activity and bone resorption [43]. PPI may inhibit Atp6V0d2, creating a scenario similar to Atp6V0d2-/- mice, which reduces mature OCs, thus inhibiting bone resorption, which seems favorable for bone formation; however, it doesn’t affect OC differentiation. These results suggest that PPIs might have a direct deleterious effect on bone cells, with the possibility of decreased bone turnover [18]. The interaction between RANK ligand and RANK receptor promotes OC activity and survival, leading to increased resorption of bone. To balance this mechanism, the protein OPG is secreted from OB [50]. OPG decreases the interaction with the RANK and diminishes bone resorption. PTH stimulates the production of RANKL and represses the expression of OPG, an effect thought to increase the activity of OCs [52]. The role of estrogen is quite important in osteoporosis. In ovariectomized rats, the relative ratio of RANKL/OPG was increased in the group fed with a low-calcium and PPI (omeprazole-30 mg/kg) diet. The bone formation marker osteocalcin was decreased, and bone resorption markers, CTX-1 levels in serum [56].
Though PPIs are not a target-specific drug, they randomly block H+ ion secretion and transportation via different mechanisms, which we have already described. By increasing pH, osteoclastic cells lose their working potential, and various biomolecules are activated due to high pH, which introduces osteoporosis in patients with prolonged PPI administration [60]. PPI administration reduces absorption of essential nutrients; PPIs majorly act on gastric secretion and reduce B12 absorption [66]. Deficiency of B12 induces OC’s maturation and formation, helping in osteoporosis induction [67]. VB12 has no direct effect on OBs, OCs, and elevated amounts of homocysteine and MMA, which may be a result of B12 deficiency-induced osteoclast genesis as well as osteoporosis by increasing bone resorption [68]. Low-iron diet, along with PPI-consuming individuals and rats, suffer from iron deficiency problems, which increase the PTH secretion [70]. PPI may regulate bone formation and resorption via PTH-rp via iron regulation.
PPI increases gastric acid pH and eventually cause hypochlorhydria, which reduces calcium ionization as well as intestinal absorption. The calcium deficiency, along with vitamin D, induces secondary hyperparathyroidism. The active hormonal form of vitamin D, i.e., calcitriol, inhibits PTH synthesis [77]. PPI therapy induces bone resorption in two ways: one via primary hyperparathyroidism through hypergastrinemia or parathyroid hyperplasia, and another via secondary hyperparathyroidism, characterized by low calcium absorption, disrupted calcium balance in circulation, lowering the plasma [Ca++], etc. PTH overexpression is the result of primary and secondary hyperparathyroidism, which reduces bone strength and subsequently induces osteoporosis. PPIs also have a deleterious effect on osteoclast like osteoblast cells. It reduces expression of genes like c-myc, c-src, TRAP, and cathepsin K in PBMC culture with 10-5M 10-3M omeprazole treatment for 21 days [18, 79]. Future studies should work on data-based studies obtained from patients and cohort studies, and focus on PPI interaction with other drugs, the impact of PPI on premenopausal and post menopause women, PPI impact on various hormones and nutrients such as PTH and OPG, vitamin D, vitamin B12, iron, and calcium to see whether it influences osteoporotic changes. Conclusively, there are several aspects of PPI that need to be studied both experimentally and theoretically, which can be impactful on preventing or treating osteoporosis. Antiresorptive agents need to be produced and studied more in order to prevent osteoporosis caused either by PPI, other drugs, or due to age.
Author Contributions
All authors contributed equally and gave final approval of the article to be published.
Conflicts of Interest
The authors declare no conflict of interest for this article.
References
- Maes ML, Fixen DR, Linnebur SA. Adverse Effects of Proton-Pump Inhibitor Use in Older Adults: A Review of The Evidence. Ther Adv Drug Saf. 2017;8(9):273-97.
- Shin JM, Kim N. Pharmacokinetics and Pharmacodynamics of the Proton Pump Inhibitors. J Neurogastroenterol Motil. 2013;19(1):25-35.
- Schubert ML, Peura DA. Control of Gastric Acid Secretion in Health and Disease. Gastroenterology. 2008;134(7):1842-60.
- Lazarus B, Chen Y, Wilson FP, et al. Proton Pump Inhibitor Use and the Risk of Chronic Kidney Disease. JAMA Intern Med. 2016;176(2):238-46.
- Myers RP, McLaughlin K, Hollomby DJ. Acute interstitial nephritis due to omeprazole. Am J Gastroenterol. 2001;96(12):3428-31.
- Geevasinga N, Coleman PL, Webster AC, eta al. Proton pump inhibitors and acute interstitial nephritis. Clin Gastroenterol Hepatol. 2006;4(5):597-604.
- Geevasinga N, Coleman PL, Webster AC, eta al. Proton pump inhibitors and acute interstitial nephritis. Clin Gastroenterol Hepatol. 2006;4(5):597-604.
- Targownik LE, Lix LM, Metge CJ, eta al. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ. 2008;179(4):319-26.
- Kaye JA, Jick H. Proton pump inhibitor use and risk of hip fractures in patients without major risk factors. Pharmacotherapy. 2008;28(8):951-9.
- Sheikh MS, Santa Ana CA, Nicar MJ, et al. Gastrointestinal absorption of calcium from milk and calcium salts. N Engl J Med. 1987;317(9):532-6.
- Leontiadis GI, Moayyedi P. Proton pump inhibitors and risk of bone fractures. Curr Treat Options Gastroenterol. 2014;12(4):414-23.
- Vangala C, Niu J, Lenihan CR, et al. Proton Pump Inhibitors, Histamine-2 Receptor Antagonists, and Hip Fracture Risk among Patients on Hemodialysis. Clin J Am Soc Nephrol. 2018;13(10):1534-41.
- Yang YX, Lewis JD, Epstein S, et al. Long-Term Proton Pump Inhibitor Therapy and Risk of Hip Fracture. JAMA. 2006;296(24):2947-53.
- Corley DA, Kubo A, Zhao W, et al. Proton Pump Inhibitors and Histamine-2 receptor Antagonists are Associated with Hip Fractures Among At-Risk Patients. Gastroenterology. 2010;139(1):93-101.
- Thong BKS, Ima-Nirwana S, Chin KY. Proton Pump Inhibitors and Fracture Risk: A Review of Current Evidence and Mechanisms Involved. Int J Environ Res Public Health. 2019;16(9):1571.
- Hussain S, Siddiqui AN, Habib A, et al. Proton Pump Inhibitors’ Use and Risk of Hip Fracture: A Systematic Review and Meta-Analysis. Rheumatol Int. 2018 Nov;38(11):1999-2014.
- Freedberg DE, Kim LS, Yang YX. The Risks and Benefits of Long-term Use of Proton Pump Inhibitors: Expert Review and Best Practice Advice From the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-15.
- Costa-Rodrigues J, Reis S, Teixeira S, et al. Dose-Dependent Inhibitory Effects of Proton Pump Inhibitors on Human Osteoclastic and Osteoblastic Cell Activity. FEBS J. 2013;280(20):5052-64.
- Ngamruengphong S, Leontiadis GI, Radhi S, et al. Proton Pump Inhibitors and Risk of Fracture: A Systematic Review and Meta-Analysis of Observational Studies. Am J Gastroenterol. 2011;106(7):1209-18.
- Ye X, Liu H, Wu C, et al. Proton Pump Inhibitors Therapy and Risk of Hip Fracture: A Systematic Review and Meta-Analysis. Eur J Gastroenterol Hepatol. 2011;23(9):794-800.
- Hyun JJ, Chun HJ, Keum B, et al. Effect of Omeprazole on the Expression of Transcription Factors in Osteoclasts and Osteoblasts. Int J Mol Med. 2010;26(6):877-83.
- Yamasaki Y, Fujimura T, Oyama K, et al. Effects of Rabeprazole on Bone Metabolic Disorders in a Gastrectomized Rat Model. Biomed Rep. 2016;5(1):118-24.
- Datta HK, Ng WF, Walker JA, et al. The Cell Biology of Bone Metabolism. J Clin Pathol. 2008;61(5):577-87.
- Sims NA, Gooi JH. Bone Remodeling: Multiple Cellular Interactions Required for Coupling of Bone Formation and Resorption. Semin Cell Dev Biol. 2008;19(5):444-51.
- Downey PA, Siegel MI. Bone Biology and the Clinical Implications for Osteoporosis. Phys Ther. 2006;86(1):77-91.
- Goltzman D, Mannstadt M, Marcocci C. Physiology of the Calcium-Parathyroid Hormone-Vitamin D Axis. Front Horm Res. 2018;50:1-13.
- Chen X, Wang Z, Duan N, et al. Osteoblast-Osteoclast Interactions. Connect Tissue Res. 2018;59(2):99-107.
- Hagymási K, Müllner K, Herszényi L, et al. Update on the Pharmacogenomics of Proton Pump Inhibitors. Pharmacogenomics. 2011;12(6):873-88.
- Tamma R, Zallone A. Osteoblast and Osteoclast Crosstalks: From OAF to Ephrin. Inflamm Allergy Drug Targets. 2012;11(3):196-200.
- Zhao C, Irie N, Takada Y, et al. Bidirectional ephrinB2-EphB4 Signaling Controls Bone Homeostasis. Cell Metab. 2006;4(2):111-21.
- Lacey DL, Erdmann JM, Shima M, et al. Interleukin 4 Enhances Osteoblast Macrophage Colony-Stimulating Factor, but not Interleukin 6, Production. Calcif Tissue Int. 1994;55(1):21-8.
- Graves DT, Jiang Y, Valente AJ. The Expression of Monocyte Chemoattractant Protein-1 and Other Chemokines by Osteoblasts. Front Biosci. 1999;4:D571-80.
- Sambandam Y, Blanchard JJ, Daughtridge G, et al. Microarray Profile of Gene Expression During Osteoclast Differentiation in Modelled Microgravity. J Cell Biochem. 2010;111(5):1179-87.
- Vishvakarma NK, Singh SM. Augmentation of Myelopoiesis in a Murine Host Bearing a T Cell Lymphoma Following in Vivo Administration of Proton Pump Inhibitor Pantoprazole. Biochimie. 2011;93(10):1786-96.
- Fata JE, Kong YY, Li J, et al. The Osteoclast Differentiation Factor Osteoprotegerin-Ligand is Essential for Mammary Gland Development. Cell. 2000;103(1):41-50.
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337-42.
- Udagawa N, Takahashi N, Yasuda H, et al. Osteoprotegerin Produced by Osteoblasts is an Important Regulator in Osteoclast Development and Function. Endocrinology. 2000;141(9):3478-84.
- Hosogaya S, Yatomi Y, Nakamura K, et al. Measurement of Plasma Lysophosphatidic acid Concentration in Healthy Subjects: Strong Correlation with Lysophospholipase D Activity. Ann Clin Biochem. 2008;45(Pt 4):364-8.
- Sims SM, Panupinthu N, Lapierre DM, et al. Lysophosphatidic Acid: A Potential Mediator of Osteoblast-Osteoclast Signaling in Bone. Biochim Biophys Acta. 2013;1831(1):109-16.
- Shah NH, LePendu P, Bauer-Mehren A, et al. Proton Pump Inhibitor Usage and the Risk of Myocardial Infarction in the General Population. PLoS One. 2015 Jun 10;10(6):e0124653.
- Mistry SD, Trivedi HR, Parmar DM, Dalvi PS, Jiyo C. Impact of proton pump inhibitors on efficacy of clopidogrel: Review of evidence. Indian J Pharmacol. 2011 Apr;43(2):183-6.
- TREMOLIZZO L., SALA G., FERRARESE C. Platelet Activation. In: Stolerman I.P. (eds) Encyclopedia of Psychopharmacology., 2010.
- Boucharaba A, Serre CM, Grès S, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest. 2004;114(12):1714-25.
- Wu H, Xu G, Li YP. Atp6v0d2 is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. J Bone Miner Res. 2009;24(5):871-85.
- Lee SH, Rho J, Jeong D, et al. v-ATPase V0 Subunit d2-deficient Mice Exhibit Impaired Osteoclast Fusion and Increased Bone Formation. Nat Med. 2006;12(12):1403-9.
- Rodan GA, Martin TJ. Therapeutic Approaches to Bone Diseases. Science. 2000;289(5484):1508-14.
- Zelzer E, Olsen BR. The Genetic Basis for Skeletal Diseases. Nature. 2003;423(6937):343-8.
- Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N Engl J Med. 2004;351(27):2839-49.
- Francis MJ, Lees RL, Trujillo E, et al. ATPase Pumps in Osteoclasts and Osteoblasts. Int J Biochem Cell Biol. 2002;34(5):459-76.
- Kearns AE, Khosla S, Kostenuik PJ. Receptor Activator of Nuclear Factor kappaB ligand and Osteoprotegerin Regulation of Bone Remodeling in Health and Disease. Endocr Rev. 2008;29(2):155-92.
- Anderson DM, Maraskovsky E, Billingsley WL, et al. A Homologue of the TNF Receptor and its Ligand Enhance T-cell Growth and Dendritic-Cell Function. Nature. 1997;390(6656):175-9.
- Silva BC, Bilezikian JP. Parathyroid Hormone: Anabolic and Catabolic Actions on the Skeleton. Curr Opin Pharmacol. 2015;22:41-50.
- Felsenfeld AJ, Levine BS. Calcitonin, the Forgotten Hormone: Does it Deserve to be Forgotten? Clin Kidney J. 2015;8(2):180-7.
- Almeida M, Laurent MR, Dubois V, et al. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol Rev. 2017;97(1):135-187.
- Streicher C, Heyny A, Andrukhova O, et al. Estrogen Regulates Bone Turnover by Targeting RANKL Expression in Bone Lining Cells. Sci Rep. 2017;7(1):6460.
- Joo MK, Park JJ, Lee BJ, et al. The Effect of a Proton Pump Inhibitor on Bone Metabolism in Ovariectomized Rats. Mol Med Rep. 2013;7(4):1267-72.
- Li YX, Chen FC, Liu T, et al. Pantoprazole (PPZ) Inhibits RANKL-Induced Osteoclast Formation and Function In Vitro and Prevents Lipopolysaccharide- (LPS-) Induced Inflammatory Calvarial Bone Loss In Vivo. Stem Cells Int. 2020;2020:8829212.
- SHIN JM., HOMERUN M., DOMAGALA F., FICHEUX H., SACHS G. Characterization of the inhibitory activity of tenatoprazole on the gastric H+,K+ -ATPase in vitro and in vivo. Biochem Pharmacol., 2006, 71 : 837-49.
- Baron R, Neff L, Louvard D, et al. Cell-Mediated Extracellular Acidification and Bone Resorption: Evidence for a Low pH in Resorbing Lacunae and Localization of a 100-kD Lysosomal Membrane Protein at the Osteoclast Ruffled Border. J Cell Biol. 1985;101(6):2210-22.
- Mattsson JP, Väänänen K, Wallmark B, et al. Omeprazole and Bafilomycin, Two Proton Pump Inhibitors: Differentiation of Their Effects on Gastric, Kidney and Bone H(+)-Translocating ATPases. Biochim Biophys Acta. 1991;1065(2):261-8.
- Ravesloot JH, Eisen T, Baron R, et al. Role of Na-H Exchangers and Vacuolar H+ Pumps in Intracellular pH Regulation in Neonatal Rat Osteoclasts. J Gen Physiol. 1995;105(2):177-208.
- Sakai D, Tong HS, Minkin C. Osteoclast Molecular Phenotyping by Random cDNA Sequencing. Bone. 1995;17(2):111-9.
- Kornak U, Kasper D, Bösl MR, et al. Loss of the ClC-7 Chloride Channel Leads to Osteopetrosis in Mice and Man. Cell. 2001;104(2):205-15.
- McColl KE. Effect of Proton Pump Inhibitors on Vitamins and Iron. Am J Gastroenterol. 2009;104 Suppl 2:S5-9.
- Lodato F, Azzaroli F, Turco L, et al. Adverse Effects of Proton Pump Inhibitors. Best Pract Res Clin Gastroenterol. 2010;24(2):193-201.
- Dai Z, Koh WP. B-vitamins and bone health–a review of the current evidence. Nutrients. 2015;7(5):3322-46.
- Kim GS, Kim CH, Park JY, et al. Effects of Vitamin B12 on Cell Proliferation and Cellular Alkaline Phosphatase Activity in Human Bone Marrow Stromal Osteoprogenitor Cells and UMR106 Osteoblastic Cells. Metabolism. 1996;45(12):1443-6.
- Vaes BL, Lute C, Blom HJ, et al. Vitamin B(12) Deficiency Stimulates Osteoclastogenesis Via Increased Homocysteine and Methylmalonic Acid. Calcif Tissue Int. 2009;84(5):413-22.
- Jensen RT. Consequences of Long-Term Proton Pump Blockade: Insights from Studies of Patients with Gastrinomas. Basic Clin Pharmacol Toxicol. 2006;98(1):4-19.
- Sharma VR, Brannon MA, Carloss EA. Effect of Omeprazole on Oral Iron Replacement in Patients with Iron Deficiency Anemia. South Med J. 2004;97(9):887-9.
- Jüppner H. Role of parathyroid hormone-related peptide and Indian hedgehog in skeletal development. Pediatr Nephrol. 2000;14(7):606-11.
- Chen GQ, Wang S, Hu SY. Osteoporosis increases chondrocyte proliferation without a change in apoptosis during fracture healing in an ovariectomized rat model. Mol Med Rep. 2012;5(1):202-6.
- Al Menhali A, Keeley TM, Demitrack ES, et al. Gastrin induces parathyroid hormone-like hormone expression in gastric parietal cells. Am J Physiol Gastrointest Liver Physiol. 2017;312(6):G649-G657.
- Amizuka N, Ozawa H, Sasaki T. The biological action of parathyroid hormone-related peptide (PTHrP) and fibroblast growth factor receptor 3 (FGFR3) on bone and cartilage. Kaibogaku Zasshi. 2000;75(5):415-25.
- Chung M, Lee J, Terasawa T, et al. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(12):827-38.
- Nieves JW. Osteoporosis: the role of micronutrients. Am J Clin Nutr. 2005;81(5):1232S-9S.
- Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev. 2001;22(4):477-501.
- Yang YX. Chronic proton pump inihibitor therapy and calcium metabolism. Curr Gastroenterol Rep. 2012;14(6):473-9.
- Prause M, Seeliger C, Unger M, et al. Pantoprazole increases cell viability and function of primary human osteoblasts in vitro. Injury. 2014;45(8):1156-64.
