Melatonin has a long history of studies which confirm its ability to inhibit cancer growth. Melatonin is present in high concentrations in the mitochondria of normal cells but is likely absent from the mitochondria of cancer cells, at least when isolated from tumors harvested during the day. Herein, we hypothesize that melatonin’s absence from cancer cell mitochondria prevents these organelles from metabolizing pyruvate to acetyl coenzyme A (acetyl-CoA) due to suppression of the activity of the enzyme pyruvate dehydrogenase complex (PDC), the enzyme that catalyzes the conversion of pyruvate to acetyl-CoA. This causes cancer cells to metabolize glucose to lactate in the cytosol (the Warburg effect). Since cancer cell mitochondria can take up nighttime pineal-derived melatonin from the blood, the indoleamine predictably promotes the conversion of pyruvate to acetyl-CoA in the mitochondria during the night. Thus, while cancer cells exhibit a typical cancer phenotype during the day, at night cancer cells have a more normal cell phenotype. Via similar actions, melatonin probably overcomes the insensitivity of cancers to chemotherapies. Hopefully, the hypothetical processes proposed herein will soon be experimentally tested.
acetyl-CoA, N-acetyltransferase, oxidative phosphorylation, aerobic glycolysis, pyruvate dehydrogenase, pyruvate dehydrogenase kinase
PDC: pyruvate dehydrogenase complex; acetyl-CoA: acetyl coenzyme A; PDK: pyruvate dehydrogenase kinase; DCA: dichloroacetate; ATP: adenosine triphosphate; ROS: reactive oxygen species; AANAT: arylalkylamine N-acetyltransferase
Soon after it’s identified involvement with reproductive physiology, especially with seasonal reproduction [1], not long after melatonin’s discovery [2] investigations were initiated to determine its actions on tumor growth. The rationale for undertaking these studies actually was derived from reports that preceded the discovery of melatonin. Pinealectomy, which diminishes circulating melatonin levels (unknown to the investigators who reported the
findings), was associated with increased tumor growth in animals [3]. Melatonin as a potential oncostatic agent was first used in animals [4], but was found to be devoid of any significant cancer-inhibiting activity contrary to that of several melatonin metabolites. In humans, one of the first reports that used melatonin, in combination with radiotherapy, to successfully treat a cancer (rhabdomyosarcoma) was published [5]. These early studies were reviewed in a symposium report which includes contributions from all the key investigators working in melatonin/cancer field at the time [6].
Concurrent with investigations related to the role of melatonin in determining tumor cell growth, small peptides were being advanced as major active secretory products of the pineal gland [7–9]. Interest in these alleged pineal secretory products, however, has waned since in some cases their structure was not characterized and, moreover, they were never proven to be released from pinealocytes. In contrast, although some of the early reports yielded ambiguous results, investigations into the role of melatonin in controlling tumor growth persisted and currently is a highly active and productive subject of research [10–18]. This action of melatonin has linked it to a very wide variety of cancer types, e.g., breast, prostate, lung, cervical, colorectal, liver, melanoma, leukemia, several childhood cancers, etc. Moreover, melatonin has been shown to be involved in tumor initiation, progression and metastasis [10, 12, 13, 19–22].
Via its stimulation of antioxidant enzymes, melatonin is a powerful direct antioxidant [23, 24] while also being a direct reactive oxygen species (ROS) (also called free radicals) scavenger [25, 26]. Because partially reduced toxic oxygen species are abundantly produced in the mitochondria, soon after melatonin’s radical quenching activities were uncovered, scientists began to investigate the actions of melatonin as a participant in mitochondrial physiology. In these investigations, melatonin improved the efficiency of the mitochondrial electron transport chain and enhanced adenosine triphosphate (ATP) production (in normal cells) [27]. Meanwhile, numerous investigators have documented the ability of melatonin to reduce ROS-mediated oxidative damage under many different experimental conditions [28–31] and in some clinical situations as well [32, 33]. Given the high production of ROS in mitochondria, one implication of the strong protection against ROS provided by melatonin was that the indoleamine may have a special relationship with mitochondria. This association was confirmed when Jou and coworkers reported that toxin-induced mitochondrial ROS, identified by immunocytochemistry, were quickly neutralized when the challenged cells were incubated in a medium containing melatonin [34, 35]. Also, in these studies, the ability of melatonin to limit ROS in mitochondria was significantly better than that provided by equivalent concentrations of commonly-used antioxidants, e.g., vitamin E. The special association that melatonin has with mitochondria was noted in several reviews published at that time [27, 36].
Based on the continuing studies reporting the existence of melatonin in many tissues [37–39], Venegas et al. fractionated brain (neurons and glia) and liver cells into several components and then measured the individual concentrations of melatonin in the isolated organelles [40]. They observed that much higher levels (up to 10-fold) of melatonin were identified in the mitochondria compared to concentrations in the nuclei, cytosol, and membranes. Additionally, the highly-elevated concentrations of melatonin in mitochondria were not diminished when the cells had been recovered from organs of long-term pinealectomized animals. This was strong evidence that mitochondrial melatonin was not a result of its uptake of pineal-derived melatonin from the circulation.
Not long after the report of melatonin in the mitochondria of normal brain and liver cells, we proposed that mitochondria of every eukaryotic cell may produce melatonin [41] (Figure 1). This was not only based on the measured levels of melatonin in mitochondria [40], but also on the evaluation of prokaryotes, which appeared on Earth about 3.5 billion years ago. Bacteria (prokaryotes) contain, and possibly synthesize melatonin [42], indicating that melatonin came into existence between 3.5 and 2.5 billion years ago. Subsequently, bacteria were engulfed and digested by early eukaryotes for their nutrient value. Eventually, the ingested bacteria established a symbiotic association with their eukaryotic hosts and evolved into mitochondria [43]; this is referred to as the endosymbiotic theory of the origin of mitochondria. In doing so, the melatonin-producing capability of the prokaryotes was presumably retained because it was a major benefit for the eukaryotes. All cells of all extinct and currently surviving species evolved from these primitive eukaryotes; during this process, mitochondria have been persevered in every cell (few exceptions) of all multicellular organisms and, as predicted, may have retained their capability to produce melatonin [43–45].
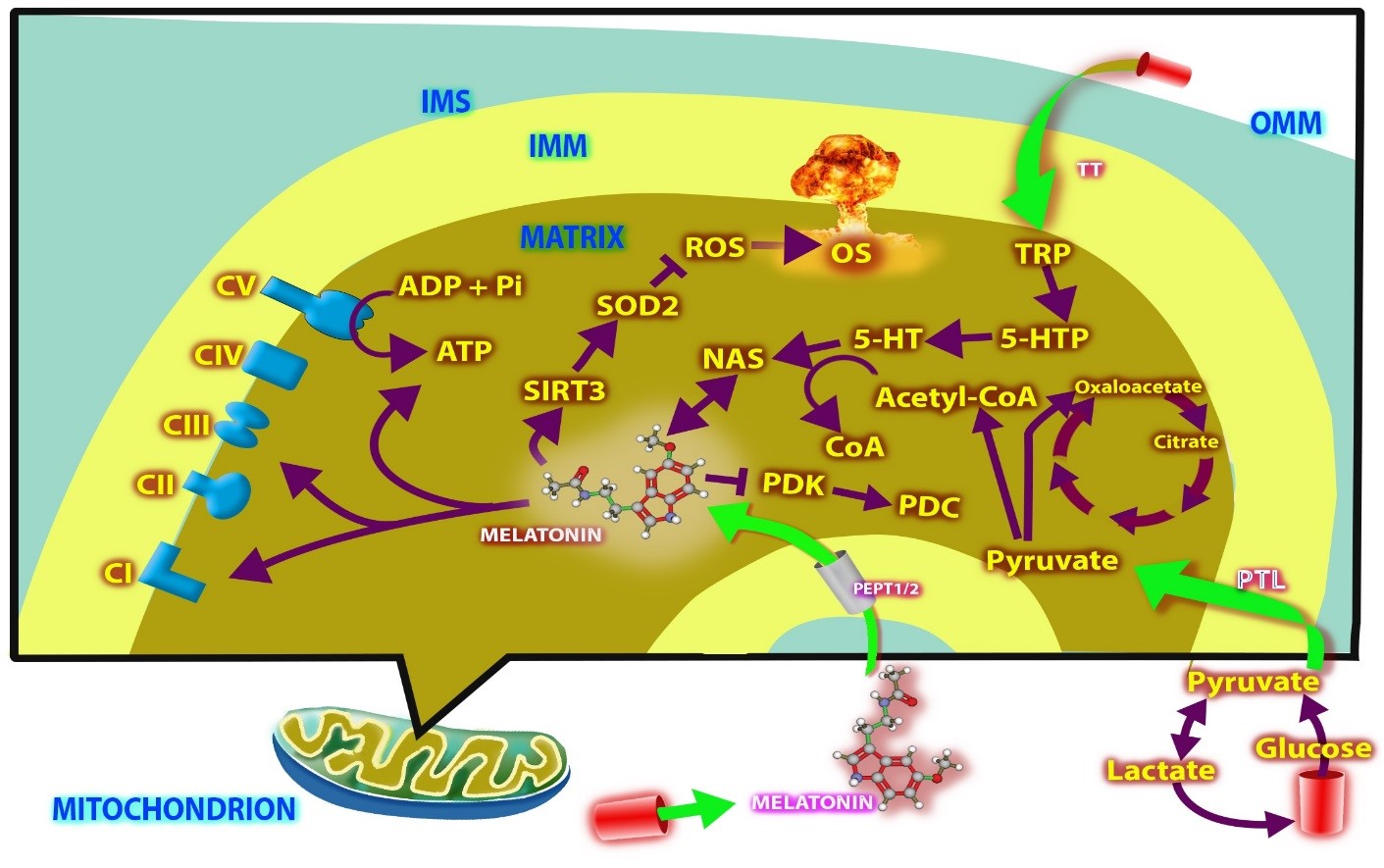
Figure 1: Normal cell mitochondria intrinsically synthesize melatonin. This occurs because pyruvate, taken into mitochondria from the cytosol, is converted to acetyl-CoA. Acetyl-CoA feeds the citric acid cycle which contributes to the efficacy of oxidative phosphorylation and mitochondrial ATP production. Additionally, after its synthesis, acetyl-CoA serves as a critical co-factor for the enzyme arylalkylamine N-acetyltransferase (AANAT) which is the rate limiting enzyme in melatonin synthesis and converts serotonin (5-HTP) to N-acetylserotonin (NAS). The synthesis of acetyl-CoA is controlled by the enzyme pyruvate dehydrogenase complex (PDC) which can be inhibited by pyruvate dehydrogenase kinase (PDK) which then prevents acetyl-CoA production. Since in the mitochondria of cancer cells, PDK is upregulated, which leads to a pronounced suppression of acetyl-CoA synthesis, cancer cells are unable to produce melatonin. In lieu of the conversion of pyruvate to acetyl-CoA in cancer cells, pyruvate is retained in the cytosol where it is used for the synthesis of cytosolic ATP and its conversion to lactate; this is referred to as the Warburg effect and is a characteristic of many solid tumors. The Warburg effect allows cancer cells to proliferate rapidly, avoid apoptosis and quickly acquire tissue biomass which also aids in metastasis. We predict that melatonin is an endogenous inhibitor of PDK allowing for acetyl-CoA production. Since normal cells synthesize melatonin, they are assured of maximal oxidative phosphorylation and a reduction in cytosolic glycolysis (Warburg effect). Since cancer cell mitochondria cannot generate their own melatonin, cytosolic aerobic glycolysis occurs which affords cancer cells physiological advantages. However, the nocturnal rise in pineal-derived blood melatonin is taken up by the mitochondria of cancer cells and inhibits PDK allowing acetyl-CoA production, melatonin synthesis and interruption of the Warburg effect. This is supported by published findings (Blask et al., 2014). This implies that breast cancer cells are neoplastic during the day and have a more normal phenotype at night, i.e., they are only part-time cancer cells. TT = tryptophan transporter; TRP = tryptophan; 5-HTP = 5-hydroxytryptophan; SODZ = manganese superoxide dismutase; ROS = reactive oxygen species; OS = oxidative stress; ADP = adenosine diphosphate; Pi = inorganic phosphate; PepT1 = oligopeptide transporters; PTL = pyruvate translocase; OMM = outer mitochondrial membrane; IMS = intermembrane spaces; IMM = inner mitochondrial membrane.
Although there is one early report claiming that pinealocyte mitochondria may produce melatonin [46], more definitive evidence for this has only recently come to light. Using the mouse oocyte, He et al. isolated the mitochondria and identified, using immunocytochemistry [47], the rate limiting enzyme in melatonin production, arylalkylamine N-acetyltransferase (AANAT), in these organelles, as had Kerenyi et al. 40 years earlier [46]. Importantly, He et al. took their study a step further and showed that purified oocyte mitochondria produced and released melatonin when these organelles were incubated in a serotonin-containing medium; serotonin is the precursor of melatonin. The results are of special importance. All mitochondria of all eukaryotic cells are descendants of the maternal supply of mitochondria, i.e., from the oocyte. Thus, since oocyte mitochondria produce melatonin it is likely that every cell in an organism has retained this ability [47].
When mitochondria were isolated from the brain of adult mice, they also were found to be capable of melatonin synthesis. Suofu et al. purified non-synaptosomal mitochondria and confirmed that they contained the protein of both enzymes required for the conversion of serotonin to melatonin, i.e., AANAT and N-acetylserotonin methyltransferase (ASMT) [48]. Also, when the purified mitochondria were incubated with deuterated serotonin, melatonin and several melatonin metabolites were formed.
Yang et al. tested whether melatonin impacted early embryonic porcine development (4- and 8-cell zygotes and the blastocyst) and found that the knockdown of AANAT, which limits melatonin synthesis, significantly impeded embryonic development [49]. They also confirmed the expression of AANAT protein in the mitochondria. The embryonic developmental slow down resulting from AANAT knockdown was reversed by supplementing the zygotes/blastocysts with melatonin. This emphasizes again that all cells in the resulting adult organism, which have their origin from the blastocyst, likely possess mitochondria with melatonin-synthesizing capability.
Because melatonin is produced in mitochondria does not mean it also cannot be taken up by these organelles after its extraction from the blood. As already mentioned, many earlier studies indicated that the incubation of cells with melatonin alters their mitochondrial physiology [34, 35]. Until recently, there was only speculation how the uptake of melatonin by mitochondria was achieved. Huo et al. found that cancer cell mitochondria possess specific transports (oligopeptide transporters, Pept1/2) which ensure and govern the rapid uptake and high concentrations of melatonin in mitochondria [50]. The presence of these transporters is consistent with the very high levels of melatonin in mitochondria after animals are given supplementary melatonin [51].
Glucose is handled differently in normal and cancer cells. In normal cells even under aerobic conditions, glycolysis results in the conversion of glucose to pyruvate in the cytosol. Pyruvate then enters the mitochondria and under the influence of PDC, it is irreversibly converted to acetyl-CoA which provides a major fuel for the citric acid cycle by coupling, via a condensation reaction, with oxaloacetate. In many solid tumor cells (and some diseases of other tissues as well), pyruvate is prevented from forming acetyl-CoA because the enzyme responsible for this, PDC, is inhibited by another enzyme, pyruvate dehydrogenase kinase (PDK). Under these conditions, mitochondrial ATP production is compromised and it is left to cytosolic glycolysis to generate ATP. This phenomenon was discovered in cancer cells decades ago and is named the Warburg effect after the individual who identified it [52].
By relegating glucose metabolism to the cytosol and abandoning mitochondrial oxidative phosphorylation cancer cells are provided an advantage for the production of macromolecules to build biomass and for the cells to escape apoptosis and undergo metastases. Cells take up large amounts of melatonin which is metabolized to several byproducts which also may impact mitochondrial physiology [53].
Melatonin, an endogenously produced molecule, has been repeatedly proven to inhibit many experimental cancer types in multiple organs [10, 12, 13, 17, 22, 54]. Numerous molecular mechanisms have been proposed to explain the ability of melatonin to resist the growth of the tumors investigated, e.g., suppression of growth factor uptake [55], estrogen inhibition [56], toll-like receptor inhibition [57], promotion of antioxidant activity [58], reduction of tumor angiogenesis [59, 60], attenuation of the HER2 signaling pathway [61], etc.
Blask et al. recently reported that in vivo breast cancer xenografts grown in rats exhibited higher metabolic activity in the day than at night. This difference was related to circulating melatonin concentrations; at night blood melatonin levels are higher and have been shown to depress tumor cell metabolism [62]. At night in these tumor xenografts, melatonin reduced glucose, lactate release, DNA synthesis and total DNA content. Also, breast cancer xenografts that are insensitive to the chemotherapies, tamoxifen or doxorubicin, rapidly become re-sensitized to the drug when concurrent melatonin treatment is initiated [63]. The mechanisms by which melatonin restores the efficacy of chemotherapies to inhibit cancer growth have not been satisfactorily resolved [63, 64].
The inhibition of pyruvate metabolism in mitochondria of cancer cells resulting in exaggerated glycolysis is well documented [65]. The mechanisms that restrict pyruvate conversion to acetyl-CoA in mitochondria relates to the reduced activity of the enzyme PDC, which in cancer cells is due to inhibition of its gatekeeper enzyme, PDK (Figure 1); PDK is usually activated in solid tumors thereby strongly inhibiting PDC. Herein, the authors hypothesize that melatonin inhibits PDK allowing for the upregulation of PDC and the conversion of pyruvate to acetyl-CoA in mitochondria. This reduces cytosolic glycolysis (the Warburg effect) and shifts glucose metabolism to the mitochondria, as occurs in normal cells [66–69].
These actions of melatonin in cancer cells are reminiscent of those caused by a class of drugs referred to as glycolytic blockers (or glycolytic inhibitors). For example, melatonin application to cancer cells causes a greatly reduced level of lactate produced by tumors suggesting a shutdown of cytosolic glycolysis [62]. The results of the actions of melatonin and the PDK inhibitors such as dichloroacetate (DCA) are similar, indicating they may use the same mechanism, i.e., an inhibition of PDK [70]. DCA is a prototypic, small molecule PDK inhibitor and has been widely tested as an anticancer agent [71].
If melatonin is a glycolysis inhibitor and works by reducing the ability of PDK to limit PDC activity as we propose, it is necessary to explain how endogenously-produced melatonin in mitochondria does not inhibit PDK and allow PDC to synthesize acetyl-CoA in cancer cells. To date, melatonin synthesis has only been documented in the mitochondria isolated from normal cells; there are no equivalent studies on cancer cell mitochondria. We propose that the mitochondria of cancer cells are incapable of synthesizing their own melatonin, at least during the day when pineal-derived blood melatonin levels are low. The reason we presume this is as follows: when pyruvate is not converted to acetyl-CoA in mitochondria, such as occurs in cancer cells, a deficiency of acetyl-CoA also develops since pyruvate is its precursor. Acetyl-CoA, however, is a necessary cofactor for the rate-limiting enzyme in melatonin production, AANAT (Figure 1); so a secondary effect of the absence of pyruvate conversion to acetyl-CoA in cancer cell mitochondria is the loss of the ability of these organelles to form melatonin. A corollary of this is that, we feel that when studies to identify melatonin in cancer cell mitochondria are attempted, it will not be found or will be there in greatly reduced amounts compared to those in normal cells, especially during the day.
While cancer cell mitochondria may be incapable of synthesizing melatonin, they are still seemingly equipped to extract it from the blood via specific transporters as noted above (Pept1/2) (Figure 1). When cancer cells are exposed to melatonin, they quickly convert to the metabolic phenotype of normal cells at least based on their glucose metabolism [64]. This we believe occurs because melatonin inhibits PDK (like DCA) in the mitochondria allowing pyruvate conversion to acetyl-CoA which then fuels AANAT. Interestingly, the authors who studied the uptake of melatonin by cancer cell mitochondria assumed that this effect accounted for the oncostatic action of the indoleamine although they proposed no mechanism by which this might be achieved [50].
Many cancers stop responding to the chemotherapies used to kill them; cancer insensitivity is highly problematic clinically since it deprives the physician of an important treatment paradigm. In a published paper, the authors observed that melatonin treatment of rats bearing human breast cancer xenografts that were unresponsive to the anticancer agent, tamoxifen, regained sensitivity to the drug (64). Thus, melatonin interrupted the insensitivity of the xenografts and reprogrammed glucose metabolism so the mitochondria again became involved in glucose metabolism with a shutdown of cytosolic glycolysis. We feel that this switch was accomplished due to melatonin’s inhibitory action on PDK which allowed the synthesis of acetyl-CoA due to the stimulation of PDC. This is similar to how other glycolytic mimetics function in reducing cancer insensitivity to chemotherapies.
The high concentrations of melatonin in the mitochondria of normal cells is presumably not shared by these organelles of cancer cells. We hypothesize that melatonin is essentially absent from cancer cell mitochondria, especially during the day, because pyruvate is not converted to acetyl-CoA, a necessary cofactor for the rate-limiting enzyme, AANAT, in melatonin synthesis. We also predict that melatonin normally inhibits PDK, the gatekeeper of PDC, thereby allowing acetyl-CoA production. In the absence of melatonin, cancer cell mitochondria continue to metabolize glucose to lactate in the cytosol, i.e., they exhibit the Warburg effect.
Cancer cells can, however, extract melatonin from the blood when the concentrations of pineal-derived melatonin are elevated. This results in the stimulation of PDC allowing cells to convert to a more normal cell phenotype by abandoning cytosolic glycolysis during the night. During the day, when blood melatonin levels drop, the cells revert back to the cancer phenotype and upregulate cytosolic glycolysis with high lactate production. In a sense, this means that breast cancer cells express a more neoplastic phenotype during the day, while at night, because of the presence of melatonin, they assume a more normal cell phenotype. Melatonin also interrupts the insensitivity of cancer cells to chemotherapies by allowing mitochondria to convert pyruvate to acetyl-CoA, thereby suppressing cytosolic glycolysis.
While the processes proposed herein are not yet confirmed by definitive experimental findings, they are consistent with the known actions of melatonin in mitochondria. One goal of this review is to stimulate interest in the functional role of melatonin in mitochondria, which the authors feel will be very broad and metabolically highly important in both normal and cancer cells. Also, there are other functions in cancer cells that are altered by melatonin that may relate to its inhibitory actions on tumor cell metabolism [72].
