Diabetes mellitus (DM) is a metabolic disorder with a variety of causes, characterized by chronic hyperglycemia and the disruption of carbohydrate, protein, and fat metabolism [1]. These conditions arise from defects in the secretion of insulin, the action of insulin, or both [1, 2]. In addition to the increased risk of microvascular complications, macrovascular complication risks are also extensively reported [2–5]. Approximately 19.8 million people are affected by the disease on the African continent [6], and 69.2% of cases go undiagnosed, mostly due to inadequate health systems, the slow progression of the disease, and little awareness of the disease within the population [7, 8]. Type 2 diabetes mellitus (T2DM) makes up 90% of all diabetes cases on the continent, with similar trends seen across the world [9, 10].
The use of blood levels of glycated hemoglobin (HbA1c) as a diagnostic tool was only implemented in 2009. The American Diabetes Association (2011) recommended that an HbA1c level of ≥48 mmol/mol (≥6.5%) should be used to diagnose individuals with T2DM [11]. Patients who presented with HbA1c levels between 39–46 mmol/mol (5.7–6.4%) would be classified as having an “increased risk” for not only diabetes but also for co-morbidities such as cardiovascular disease (CVD). These patients would be advised about effective strategies, such as an exercise regime and a change in diet, to lower their risk. The recommendations mentioned above were accepted by the World Health Organization (WHO) [11]. Clinicians favored this test as it is convenient, does not require fasting, and is not affected by short-term lifestyle changes [1].
In South Africa, recommendations for the diagnosis of T2DM were put forward by the Society for Endocrinology, Metabolism and Diabetes of South Africa (SEMDSA) [12]. These stipulate that symptomatic patients (frequent uptake of fluids, frequent urination, and blurred vision) who have random plasma glucose, fasting plasma glucose, and HbA1c readings greater than 11.1 mmol/L, 7.0 mmol/L, and 6.5%, respectively, can be diagnosed with T2DM [12]. For asymptomatic individuals, the random plasma glucose, fasting plasma glucose, and HbA1c readings must be the same as for symptomatic individuals; however, either of the tests must be repeated on separate days but within two weeks of the first test [12].
Biomarkers provide a quantification of a biological process [13]. Due to the nature of the disease, some patients may show symptoms such as excessive hunger, frequent urination, and consistent thirst, while others may be asymptomatic [14]. The usage of biomarkers has been shown to aid in the definitive diagnosis of various diseases, such as cancer. A variety of protein molecules have been identified to serve as biomarkers in diabetes management [15]. C-peptide is synthesized during insulin production as it is cleaved from proinsulin and stored in secretory granules before being released into the bloodstream [16–18]. C-peptide plays a vital role in the synthesis of insulin, as it links the A and B chains of the molecule, allowing the folding and formation of the interchain disulfide bonds. C-peptide is cleaved from proinsulin via proteolytic processing. After which the carboxyl-terminal of insulin’s B chain is left exposed to effectively interact with the insulin receptor [19]. Lower values of C-peptide are linked with poor glycemic control and are therefore linked with higher HbA1c values [20–22].
The glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are peptides that belong to the glucagon-secretin family and are secreted from different cells in the small intestine as a response to nutrient ingestion [23]. The presence of glucose stimulates both hormones [23]. Each of the incretin hormones has specific receptors; both are expressed in high numbers on the β-cells [24–26]. The loss of the incretin effect is one of the characteristics of T2DM [27]. The loss of action influences plasma glucose levels after meal ingestion due to the lack of insulinotropic effect, thus contributing to hyperglycemia [28]. While GIP is ineffective regardless of dose, a slight increase in GLP-1 may stimulate the secretion of insulin to the levels seen in healthy individuals [29, 30]. It was previously accepted in T2DM that the secretion of GIP is preserved, but the response is impaired. With GLP-1 in T2DM, the secretion is lower, but the action is maintained [31]. However, more recent meta-analyses show there may be no systematic differences between the secretion of the incretin hormones in healthy individuals and those with T2DM [32].
Visfatin, also known as pre-B-cell colony-enhancing factor 1 (PBEF1), is secreted mostly by visceral fat. It was reported to mimic the actions of insulin [33, 34] due to its ability to prevent glucose release from the liver, increase glucose absorption in both monocytes and adipocytes, and stimulate the increase of triglyceride synthesis [35]. It is the glucose-lowering characteristic of visfatin that has made it an attractive prospect as a novel medication for diabetes [36]. Elevated visfatin in blood plasma causes obesity, T2DM, and metabolic syndrome (MetS) [37]. Clinical studies have reported that plasma visfatin levels are increased in T2DM patients compared with healthy individuals [35–40].
Ghrelin plays a vital role in stimulating gastric acid secretion, regulating glucose and lipid metabolism, as well as modulating learning and memory functions [41–43]. Studies show that circulating ghrelin was negatively correlated with BMI, insulin resistance, waist circumference, and MetS. These findings indicated that ghrelin might be involved in the development of T2DM [44–46]. It has been reported that ghrelin positively influences glucose metabolism and insulin sensitivity [47–49]. A recent study by Li et al. [50] investigated serum ghrelin levels between T2DM patients and healthy individuals. The results of the study showed a decrease in the levels of ghrelin in the T2DM case group. The serum ghrelin levels were negatively correlated with HbA1c levels.
Plasminogen activator inhibitor-1 (PAI-1) is produced by a variety of cells, including adipocytes, hepatic cells, and endothelial cells [51, 52]. An increase in PAI-1 expression inhibits the fibrinolytic process, subsequently leading to the deposition of fibrin and tissue damage [53, 54]. A variety of inflammatory cytokines, such as TNF-α and IL-6, as well as hormones, including epinephrine and insulin, influence this peptide [55–58]. A study conducted by Nkansah et al. [59] in which 30 healthy blood donors were compared with 30 T2DM patients with poor glycemic control and 30 patients with good glycemic control showed increased PAI-1 levels in the patients with T2DM. The results indicated a statistically significant difference in the group that contained the patients with poor glycemic control. Additionally, a significant difference was observed between HbA1c and PAI-1 levels [59]. Further associations have been reported in human cross-sectional studies between increased concentrations of PAI-1 and obesity [60, 61], impaired glucose tolerance (IGT) [61, 62], insulin resistance [61, 62], and T2DM [63, 64].
Leptin is known as the ‘satiety hormone’ as it helps to inhibit hunger by regulating energy balance. It is secreted by adipocytes and is involved in controlling the food intake and leads to the suppression of appetite [65]. The action of leptin is to stimulate thermogenesis, inhibit appetite, decrease glucose levels, and reduce body weight and fat. It acts through its receptors, LEPRs, located in specific neuron populations of the brain [66]. In those who are obese, the decreased leptin sensitivity results in the body’s inability to detect satiety despite high energy stores [67]. A recent study by Ahmad [68] evaluated the concentrations of serum leptin in 30 non-T2DM participants and 30 T2DM patients. The case group showed significantly higher levels of leptin, glucose, and cholesterol compared to the control group. Additionally, the serum leptin levels positively correlated with HbA1c values [68].
Adiponectin was previously thought only to be produced and secreted by adipose tissue. However, this has since been disproved as adiponectin is expressed in human osteoblasts [69], placental tissue [70], epithelial cells [71], and myocytes [72]. Lower levels of adiponectin are associated with T2DM [73–75]. Adiponectin functions as an insulin sensitizer and exhibits anti-inflammatory and anti-diabetic properties [76]. In animal models, a decrease in adiponectin levels preceded the onset of T2DM [77]. In contrast to other adipokines, the serum levels of adiponectin are decreased in an insulin-resistant state [78].
Adipsin is one of the major proteins in adipose tissue [79]. It is the rate-limiting enzyme involved in the alternate pathway of complement activation [80]. A human study conducted by Wang et al. [81] showed that serum adipsin levels were highest in those with normoglycemia compared to individuals with known DM who had lower circulating levels. Similar results were published in a meta-analysis by Tafere et al. [82]; the plasma concentrations of adipsin in T2DM patients with IGT are lower than when compared to healthy individuals. In contrast to the mice study, adipsin levels are not associated with the function of β-cells but are negatively correlated with insulin resistance [81].
There have been numerous population-, family- and twin studies that have provided evidence on the heritability of T2DM, which ranges from 20–80% [62, 83, 84]. For individuals with one affected parent, the lifetime risk of developing T2DM is 40% [85]. The risk is greater if the mother is affected [86]. The risk increases to 70% if both parents are affected [85, 87]. There have been numerous genome-wide association studies (GWAS) conducted to better understand the genetic mechanisms at play that contribute to the development of the disease with over 400 genetic variants associated with T2DM, to provide a resource to facilitate further investigations into the understanding of biological mechanisms causing T2DM [88]. To gain a better understanding of genes associated with T2DM, this study evaluated the possible role that rs6235 in proprotein convertase subtilisin/kexin type 1 (PCSK1) and rs2208203 in proprotein convertase subtilisin/kexin type 2 (PCSK2) may play in the onset of T2DM in this population. The PCSK SNPs were selected for the reason that they are involved in the synthesis pathways of multiple biomarkers, including C-peptide, GLP-1, glucagon, GIP, and ghrelin, for a more holistic approach.
The PCSK1 gene encodes the PC1/3 protein and is located on chromosome 5q15-21 [89]. The PCSK2 is located on chromosome 20p12.1 and encodes the PC2 protein. Both PC1/3 and PC2 are selectively expressed in neuroendocrine and endocrine cells, highlighting their importance in the processing of prohormones [89–92]. Both PC1/3 and PC2 are found 115 in endocrine and neuronal cells that contain secretory granules [89–91].
These enzymes are involved in the processing of hormones involved in glucose homeostasis. Proinsulin is the precursor to insulin and is synthesized in the pancreatic β-cells. Active insulin is produced via the action of both PC1/3 and PC2; however, PC1/3 plays a more significant role [93, 94]. The processing of proinsulin yields C-peptide and insulin. Similarly, the processing of proglucagon yields glucagon and GLP-1. The processing of proglucagon, which occurs in the pancreatic α-cells, is conducted by PC2 [95–97]. The processing of the incretin hormones is also performed by PC1/3. The conversion of proglucagon to GLP-1 occurs in the intestinal L-cells and is performed solely by PC1/3, as the L-cells lack PC2 expression [95, 98], and GIP is produced in the intestinal K-cells [99]. In addition, proghrelin undergoes processing by PC1/3 to produce ghrelin, which is secreted by the A cells of the stomach [100, 101].
The aim of this study was to determine the concentrations of biomarkers associated with T2DM using enzyme-linked immunosorbent assay (ELISA). Genetic variants associated with the studied biomarkers were evaluated to determine whether disease risk could be elucidated from biomarker levels and genetic variants associated with the biomarkers in this study population.
Ethical approval for this study was obtained prior to the onset of the study from the UFS HSREC (UFS-HSD2018/1589/2901). Archived samples from study UFS-HSD2017/0979 were used. A case-control study was conducted on a cohort consisting of 78 non-Caucasian individuals of African ancestry, with ages ranging between 38 and 62 years. The control cohort consisted of individuals with an HbA1c level < 6.5% and were sampled from public areas, such as malls and community centers. The case-cohort included T2DM patients who had been previously diagnosed by a physician and with an HbA1c value > 6.5%. Informed consent was obtained from all participants before acquiring blood samples for biomarker and genetic analysis; one SST and two EDTA tubes were taken from each participant. In addition, height and weight measurements were collected to calculate the BMI of each participant.
The biomarker analysis was conducted at the Optics and Imaging Centre at the University of KwaZulu-Natal (UKZN) Nelson R. Mandela School of Medicine. Ten biochemical markers, including – C-peptide, insulin, glucagon, visfatin, resistin, PAI-1, ghrelin, leptin, GLP-1, and GIP, were screened using the Bio-Plex Pro™ 10-Plex Human Diabetes Assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA; catalogue number: #171A7001M) according to the manufacturer’s instructions. The plate was read using the Bio-Plex® 200 system, and results were captured using the Bio-Plex Manager™ software. The genetic analysis was conducted at the University of the Free State (UFS). Nucleic acid isolation was completed using the QIAGEN QIAamp® DNA Blood Mini Kit (Qiagen, Hilden Germany; lot number: 160043295 reference number: 51104). For molecular genotyping, qPCR was used with TaqMan chemistry. IDT rhAmp™ Genotyping Master Mix 2X (Integrated DNA Technologies, Inc., Coralville, IA USA; catalogue number.: 1076014), IDT rhAmp® Reporter Mix, and rhAmp® SNP Assay (primers and probes), for the SNP’s rs6235 and rs2208203 of the genes PCSK1 and PCSK2 respectively, were designed and manufactured by IDT (Integrated DNA Technologies, Inc., Coralville, IA, USA). gBlocks® Gene Fragments (Integrated DNA Technologies, Coralville, IA, USA) were used as the positive controls. Applied Biosystems™ QuantStudio® 5 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) was utilized for the qPCR. GraphPad Prism version 8.2.0 (GraphPad Software Inc. CA, USA) was used for analyzing the results obtained for both the biomarker analysis and molecular genotyping. Statistical analyses performed on the data included the Pearson correlation coefficient to measure the linear correlation between the biomarkers and their association with HbA1c and BMI and the two-sided Fisher’s exact test to determine if there was statistical significance within the study population based on the genotyping results obtained. A Chi-square test was performed to determine the differences between the frequencies of the genotypes in the population. Additionally, one-way ANOVA and Tukey’s multiple comparison tests were conducted based on the genotyping results obtained for rs2208203, as all possible genotypes were present in the study population.
The control group consisted of 15 males and 24 females (n = 39), while the T2DM case cohort consisted of 12 males and 27 females (n = 39). The average age of the control and T2DM case groups was 45.26 (± 5.93) and 51.41 (± 7.22), respectively. A p-value ≤0.05 was regarded as statistically significant. Comparison of the age between the two study cohorts yielded a value of p < 0.0001. The mean BMI of the T2DM case group was 33.69 kg/m2 (± 7.74) and was higher than the BMI of the control group, 29.14 kg/m2 (± 6.65). The T2DM case group was significantly higher than the control group, as indicated by a p-value of 0.0071. Similarly, the average HbA1c values between the two groups were also statistically significant: p-value < 0.0001. The control and T2DM case groups had average HbA1c values of 5.66% (± 0.27) and 8.97% (± 1.97), respectively. A comparison between these variables and the two cohorts is indicated in the table below (Table 1).
| Variable | Control group (SD) | Case group (SD) | p-value (* indicates p-value < 0.05) |
| Age (years) | 45.26 (± 5.93) | 51.41 (± 7.22) | < 0.0001* |
| BMI (kg/m2) | 29.14 (± 6.65) | 33.69 (± 7.74) | 0.0071* |
| HbA1c (%) | 5.66 (± 0.27) | 8.97 (± 1.97) | < 0.0001* |
Table 1: The mean age, BMI, and HbA1c values for the control and T2DM case groups. The p-values are also indicated. Statistically significant values were obtained for each of these categories.
The biomarker analysis evaluated the serum levels of 10 biomarkers. One biomarker, PAI-1, failed to register any readings. 4 of the 10 biomarkers showed statistically significant values between the two study cohorts. These biomarkers were GIP (p-value = 0.0004), glucagon (p-value = 0.0034), visfatin (p-value = 0.0009), and resistin (p-value = 0.0398). The average concentrations of each biomarker are depicted in the table (Table 2).
Pearson correlation coefficients were calculated to determine the association, or linear correlation, between biomarker concentrations with HbA1c and BMI in both groups. For HbA1c vs. biomarkers in the control group, ghrelin, GLP-1, and insulin presented with p-values ≤0.05. In comparison with BMI, only leptin presented with a statistically significant p-value of 0.015. Under the same analysis, the case group showed the comparison between the biomarkers with HbA1c and BMI values; only GLP-1 presented statistically significant values of 0.028 and 0.02, respectively.
| Biomarker | Control group (pg/ml) | Case group (pg/ml) | p-value (* indicates p-value < 0.05) |
| C-peptide | 1323.98 (1338.00) | 932.90 (1162.00) | 0.203 |
| Ghrelin | 448.79 (1198.00) | 202.83 (150.60) | 0.254 |
| GIP | 456.26 (248.80) | 231.41 (186.20) | 0.0004* |
| GLP-1 | 871.00 (1286.00) | 310.68 (209.50) | 0.1173 |
| Glucagon | 2064.46 (515.80) | 1686.33 (467.40) | 0.0034* |
| Insulin | 3.53e + 48 (5.948e + 25) | 1.292e + 50 (7.184e + 50) | 0.45 |
| Leptin | 12246.60 (15981.00) | 9501.7 (11158.00) | 0.4272 |
| Visfatin | 12389.90 (8635.00) | 5608.15 (4592.00) | 0.0009* |
| Resistin | 2.110e + 25 (5.498e + 25) | 1.141e + 22 (2.535e + 22) | 0.0398* |
Table 2: Average concentrations for each biomarker for the control and T2DM case group.
For molecular genotyping analysis, genes associated with the biosynthesis of insulin, C-peptide, GLP-1, glucagon, GIP, and ghrelin were selected. The SNPs selected were rs6235 in PCSK1 and rs2208203 in PCSK2. The molecular genotyping results of rs6235 showed that 32 individuals in the control group had the CC genotype, and 7 individuals had the CG genotype. Analysis of the T2DM case group yielded similar results, 31 individuals had the CC genotype, and 8 individuals presented with the CG genotype. A two-sided Fisher’s exact test was conducted to determine whether there was a significant difference between the T2DM-case group and the control group based on the distribution of the genotypes. A p-value ≤0.05 was deemed statistically significant. The p-value that this analysis yielded was ≥0.999. The allele frequencies and OR values for each allele for rs6235 can be viewed in the table (Table 3).
| Gene | SNP | Allele | Allele frequency: control | Allele frequency: T2DM case | Odds ratio | 95% CI | p-value |
| PCSK1 | rs6235 | C | 0.91 | 0.90 | 1.159 | 0.387-3.082 | > 0.999 |
| rs6235 | G | 0.09 | 0.10 | 0.863 | 0.325-2.587 | > 0.999 |
Table 3: Allele distribution for rs6235 in PCSK1. The table includes the allele frequencies and OR values.
Similar to the biomarkers, the SNPs were compared with HbA1c and BMI to determine whether these variables were influenced by the presented genotypes. In comparing the CC and CG genotypes with HbA1c, an insignificant p-value of 0.663 was calculated. The comparison of these genotypes with BMI resulted in a p-value of 0.443.
Correspondingly to rs6235, SNP rs2208203 also failed to show any statistical significance difference between the two study cohorts. The control group showed 34 individuals presented with the TT genotype, 5 individuals possessed the TC genotype. The T2DM case group showed all three genotypes; 33 individuals were homozygous for the reference T allele, 5 individuals were heterozygous (TC), and one individual was homozygous for the alternate C allele. A Chi-square test was conducted to determine the difference between the genotype frequencies. The p-value obtained from this analysis was 0.602. The allele frequencies and OR values for rs2208203 for this study population are shown in the table below (Table 5).
| Biomarker | CC | CG | p-value |
| C-peptide | 1147.00 | 1086.00 | 0.873 |
| Ghrelin | 371.80 | 157.30 | 0.429 |
| GIP | 345.70 | 387.00 | 0.624 |
| GLP-1 | 699.60 | 436.00 | 0.554 |
| Glucagon | 1862.00 | 1881.00 | 0.903 |
| Insulin | 1.001e + 50 | 6.710e + 48 | 0.645 |
Table 4: Average biomarker concentrations for the genotypes acquired for SNP rs6235. The biomarkers are those involved in the PCSK1 pathway.
| Gene | SNP | Allele | Allele frequency: controls | Allele frequency: T2DM cases | Odds ratio | 95% CI | p-value |
| PCSK2 | rs2208203 | T | 0.94 | 0.91 | 1.439 | 0.471-4.185 | 0.765 |
| rs2208203 | C | 0.06 | 0.09 | 0.695 | 0.239-2.124 | 0.765 |
Table 5: Allele distribution for rs2208203 in PCSK2. This table includes allele frequencies and OR values for each allele.
One-way ANOVA tests were used to determine if there was an association of the three genotypes present in this study group with HbA1c and BMI. These comparisons can be viewed in the table (Table 6). The comparison between the three genotypes and BMI showed a statistically significant p-value of 0.008. A Tukey’s multiple comparison test was conducted to show the difference between the TT vs. CC and TC vs. CC genotypes. Both these p-values were 0.006. The table illustrates the analysis of the biomarkers in which PCSK2 plays a role (Table 7). These biomarkers are C-peptide, insulin, and glucagon. No significant statistical difference was seen between these biomarkers and the genotypes of rs2208203.
| Variable | TT | TC | CC | p-value |
| HbA1c (%) | 7.261 | 7.610 | 8.00 | 0.854 |
| BMI (kg/m2) | 31.27 | 29.81 | 53.80 | 0.008 |
Table 6: Association of genotypes with average HbA1c and BMI values. A p-value < 0.05 was obtained in comparing the genotypes with associated BMI values.
| Biomarker | TT | TC | CC | p-value |
| C-peptide | 1180.00 | 931.00 | 554.60 | 0.767 |
| Insulin | 9.684e + 49 | 5.571e + 47 | 3.04e + 33 | 0.911 |
| Glucagon | 1912.00 | 1554.00 | – | 0.0698 |
Table 7: Biomarker concentrations associated with the genotypes for rs2208203.
The biomarkers GIP, glucagon, visfatin, and resistin were statistically significant between the control and T2DM case groups. However, PAI-1 failed to register any readings.
The GIP is an incretin hormone that stimulates the secretion of insulin [102]. The incretin effect in those with T2DM is impaired; however, while the action is diminished, the secretion is preserved [27]. In this study, the average value of GIP in the control group was almost double that of the T2DM case group, 456.26 pg/ml vs. 231.41 pg/ml (p-value = 0.0004). Similarly, studies conducted by Skrha et al. [103] and Vollmer et al. [104] evaluated the activity of GIP using meal tests and oral glucose tolerance test (OGTT) and found GIP levels significantly higher in the T2DM case group. Studies have shown that impairments of the incretin effect may be an early sign of β-cell dysfunction. The incretin effect is further affected when the functioning of β-cells decreases [105, 106]. The use of GIP as a biomarker in determining the progression of T2DM could be effective in identifying T2DM in its early stages.
The serum concentration of resistin in the study population was exponentially high. The mean concentration of the control group was 2.110e + 25 pg/ml, while the concentration of the T2DM case group was 1.141e + 22 pg/ml; this yielded a p-value of 0.0398. This is supported by a study conducted using OGTT to measure resistin levels before and after a 75 g glucose load. The resistin levels in the T2DM case group were significantly lower than those in the control group [107]. In contrast, the role of cytokines in the onset of T2DM showed that resistin levels were significantly increased in the T2DM case group. The role of resistin in the onset of T2DM remains uncertain. Some reports suggest that resistin levels have no effect on insulin resistance but may instead contribute to adiposity [108], while others have documented a link between insulin resistance and resistin levels [109, 110]. The conflicting reports from previous studies in relation to this one suggest that resistin should be further investigated in large cohorts correcting for obesity to determine its suitability as a biomarker in determining the onset of T2DM.
In this study, the visfatin levels were also almost double in the T2DM case group than the control group, 12 389.90 pg/ml vs. 5 608.15 pg/ml, p-value = 0.0009. Dissimilarly, Li et al. [111] and Yaturu et al. [112] reported visfatin levels that were lower in T2DM case groups than in the control groups. However, numerous other studies have reported greater visfatin levels in the T2DM cohorts [27, 113, 114]. The study conducted by Abd Rabo et al. [113] evaluated cohorts that were further divided by BMI, obese (≥ 30) and non-obese (BMI ≤ 30). The obese T2DM cohort exhibited greater visfatin concentrations than the non-obese T2DM cohort [113]. Visfatin is mostly secreted by visceral fat and has been shown to mimic the role of insulin. It is regarded as a pro-inflammatory adipokine [115]. The exact biological role of visfatin has yet to be elucidated. The significantly lower levels of visfatin seen in the T2DM case group could be attributed to the insulin therapy administered to most patients in this group. Additionally, it could be attributed to lower visceral fat levels, however, as fat distribution data was not collected in this study this cannot be confirmed. While significantly different between the two cohorts of this study, the reliability of visfatin as a biomarker in T2DM remains uncertain.
An average glucagon concentration of 2 064.46 pg/ml was measured in the control group, compared to 1 683.33 pg/ml in the T2DM case group, yielding a p-value of 0.0034. It has been documented that an α-cell in a T2DM state secretes high levels of plasma glucagon [116]; thus, the results in this study were not expected. This could be explained by the use of anti-diabetic medication or the state of fasting between the groups. Thus, unless measured during fasting, glucagon fluctuates and will not serve as a good biomarker.
PCSK1 (rs6235)
All 78 samples were genotyped for SNPs with reference numbers rs6235 and rs2208203 using quantitative polymerase chain reaction (qPCR). For SNP rs6235, the study population showed 63 CC homozygotes and 15 heterozygotes OR value of 0.863 (Table 3). The T2DM cohort contained 31 homozygotes and 8 heterozygotes. No statistical significance difference was found between the cohorts (p-value > 0.9999). The allele frequency in the study population was like that seen in African populations, with a major allele frequency (C) of 90% (Figure 1).
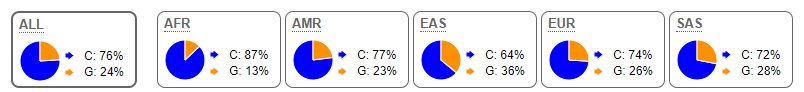
Figure 1: Allele frequency of rs6235 in various population groups (1000 Genomes Project Phase 3; Ensembl.org).
The rs6235 SNP has been associated with obesity [117]. To determine whether this could be the case in our study, unpaired t-tests were performed to determine if the BMI and HbA1c levels between those with the CC and CG genotypes were different. The CC genotype group had an average BMI of 31.71 kg/m2 (± 7.85), while the CG genotype group had an average of 30.04 kg/m2 (± 5.98). The p-value obtained was 0.443. The difference between the average HbA1c values of those with the CC 7.37% (± 2.25) and CG 7.093% (± 1.89) genotypes was investigated. The comparison obtained an insignificant p-value of 0.663. In this study population, the alternate G allele did not confer higher BMI or HbA1c values, indicative of disease progression.
To date, several GWAS have been conducted to determine whether rs6235 contributes to the risk of obesity. Studies were first conducted on European populations [117, 118] but have since been replicated in Asian [119, 120] and Mexican populations [121]. Results have varied between population groups; a study on a Swedish population showed no relationship between rs6235 and obesity [118], while a Danish population stated that rs6235 is associated with obesity [122]. In a study conducted by Choquet et al. [123], the association of SNPs in the PCSK1 gene, including rs6235, was investigated in an American population. In the African-American group, a significant association was found between the rs6235 minor allele and both obesity and BMI [123]. In a recent study conducted in France that studied the effect of PCSK1 gene variant clusters on obesity, it was found that PCSK1 gene variant clusters with a partial or neutral effect on PC1/3 activity did not have an effect on obesity [124].
Comparisons between the levels of biomarkers involved in the PCSK1 pathway (C-peptide, insulin, glucagon, ghrelin, GLP-1, and GIP) and the genotypes present in this study population were conducted to determine if the alternate G allele for rs6235 played a role in the levels of these biomarkers. The results of this analysis showed that no statistical difference was found between the two genotypes and biomarker levels in this study population (Table 4). Limited studies are available regarding the association between the presence of the PCSK1 rs6235 SNP and levels of biomarkers, with none performed on African populations. The rs6235 variant has been described in the literature to cause an increase in insulin levels [125, 126]. In a study conducted by Enya et al. [127], the PCSK1 variant was significantly associated with higher levels of fasting insulin (p = 0.034) and homeostatic model assessment for insulin resistance (HOMA-IR) (p = 0.030). Once adjustments had been made for age, BMI, and sex, there was a significant association with the development of T2DM (p = 0.0043) [127]. With regards to the relation of other biomarkers and rs6235, a study conducted on a Danish population by Gjesing et al. [128], showed that the rs6235 SNP caused higher GIP and glucagon levels during a meal-test. However, no significant differences were found in proinsulin and insulin levels in the meal test [128].
The results obtained in this study could be attributed to the small sample size (N = 78) and the use of serum biomarker levels, as, in previously mentioned studies, insulin and glucagon levels were measured using OGTT or meal tests. The PC1/3 enzyme is not the rate-limiting enzyme in the processing of proinsulin and proglucagon; therefore, the presence of the alternate allele may influence the processing of the enzyme through the entire pathway rather than the amount of the product.
PCSK2 (rs2208203)
The genotyping results of rs2208203 in the PCSK2 gene showed 67 participants were homozygous for the reference T allele. This was distributed relatively evenly between the T2DM case group (N = 33) and the control group (N = 34). As in the case of rs6235, in the PCSK1, allele frequencies observed in the study population were similar to published allele frequencies from the 1000 Genomes Project (Figure 2).
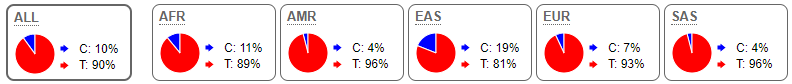 Figure 2: Allele frequencies for rs2208203 in various population groups (1000 Genomes Project Phase 3; Ensembl.org).
Figure 2: Allele frequencies for rs2208203 in various population groups (1000 Genomes Project Phase 3; Ensembl.org).
The OR test was used to determine if the presence of the alternate C allele confers greater disease risk. An OR value of 0.695 suggests this not to be the case in the study population. The results of this analysis are outlined in the table (Table 5). One-way ANOVA tests were performed to determine the relationship between the genotypes, HbA1c, and BMI. The relationship between rs2208203 and HbA1c yielded insignificant results (p-value = 0.854). However, the comparison between the genotypes and BMI produced statistically significant results, with a p-value of 0.008. A Tukey’s multiple comparison test was used to investigate this further. The results from the test showed TT vs. CC and TC vs. CC were statistically significant, p = 0.006 and p = 0.006, respectively. The results obtained for this analysis could be attributed to the individual homozygous for the alternate C allele. This individual had a BMI value of 53.80 kg/m2. In comparison, the mean BMI values for those with the TT and TC genotypes were 31.27 kg/m2 and 29.81 kg/m2, respectively. Comparison between these genotypes (TT vs. TC) yielded a p-value of 0.834. Jonsson et al. [129] found that rs2208203 had no significant effect on BMI. The result obtained in our study is primarily due to the one individual with the CC genotype having a high BMI. This could be attributed to a small sample size, as one individual presented as homozygous for the minor C allele.
The association between the genotypes of rs2208203 in PCSK2 and biomarkers in which this gene plays a role is presented in the table (Table 7). No statistical significance was found between the genotypes of rs2208203 and any of the associated biomarkers. There are limited studies available that have investigated the relationship between the PCSK2 gene SNP rs2208203, glucose homeostasis, and its contribution to T2DM. A GWAS investigated the risk of the minor/alternate allele from SNP rs2208203 on T2DM development [129]. In addition to measuring insulin and glucagon levels, gene expression was conducted in the islet cells of the pancreas. The gene expression analysis was correlated with HbA1c values, and the results showed that PCSK2 expression was negatively correlated with HbA1c values. Hyperglycemic individuals reported lower PCSK2 expression and higher HbA1c values. The proinsulin, insulin, and glucagon measurements were done during various intervals of OGTTs. The results of the study showed that the presence of the alternate allele from the SNP rs2208203 resulted in significantly lower insulin and glucagon levels [129]. The comparison of insulin and glucagon levels in our study with the presence of the alternate allele of SNP rs2208203 did not confer significantly lower insulin or glucagon levels, p = 0.094 and p = 0.0698, respectively. The differences could be attributed to the GWAS study being conducted on a Scandinavian population as well as the number of participants of more than 7,000 participants [129].
The lack of statistical significance between the three biomarker levels and the genotypes associated with rs2208203 can also be attributed to the role that PC2 plays in the processing of these prehormones; it is not a rate-limiting enzyme. Therefore, the processing of these biomarkers can be measured with tests such as OGTTs, and this may prove more beneficial in understanding the role of PCSK2 and more specifically, rs2208203 in the development of T2DM. The GWAS study reported that PCSK2 does play a role in the development of T2DM; however, the specific role of SNP rs2208203 was not investigated [129].
