Subacute sclerosing panencephalitis (SSPE) is a neurodegenerative disease caused by persisting measles virus (MeV) in the central nervous tissue of the patients. About one in ten thousand children who contract the infection in early childhood develop SSPE. Fulminant SSPE is a rare disease with fatal consequences. Magnetic resonance imaging (MRI) can help in making a diagnosis, but all the possible changes encompassing all stages and their correlation with severity are yet to be described. We report a case of fulminant SSPE with unusual findings on MRI, which led to great difficulty and delay in making the correct diagnosis.
subacute sclerosing panencephalitis, measles, vasculitis, acute disseminated encephalomyelitis, neuroimaging
SSPE: subacute sclerosing panencephalitis; MeV: measles virus; MRI: magnetic resonance imaging; GCS: Glasgow Coma Score, CSF: cerebrospinal fluid; ADEM: acute disseminated encephalomyelitis; IV: intravenous
Subacute sclerosing panencephalitis (SSPE) is a neurodegenerative disease caused by persisting measles virus (MeV) in the central nervous tissue of the patients. About one in ten thousand children who contract the infection in early childhood develop SSPE [1]. The Indian population has an annual incidence of 21 cases per million people [2]. The course of the disease usually runs over one to three years. It is said to be fulminating if the course is restricted to less than one year [3]. Fulminant SSPE is a rare disease with fatal consequences. Magnetic resonance imaging (MRI) can help in making a diagnosis, but all the possible changes encompassing all stages and their correlation with severity are yet to be described. We report a case of fulminant SSPE with unusual findings on MRI, which led to great difficulty and delay in making the correct diagnosis.
A 12 years old, developmentally normal premorbid, unimmunized male child presented to our centre with increased drowsiness and decreased oral intake for the past fifteen days. The child was also reported to have sudden jerky movements of bilateral lower limbs leading to frequent falls for the past two months. The child was conscious but not aware during the episodes. There was no loss of consciousness after these episodes of abnormal movements, with each episode lasting for around 30 seconds. There was no history of uprolling of eyeballs, clenching of teeth, deviation of mouth, or urinary incontinence during the episodes. There was no family history of a similar illness. General physical examination revealed average built with no dysmorphism or neurocutaneous markers. Neurological examination revealed normal motor and sensory system, but the sensorium was depressed with Glasgow Coma Score (GCS) E3V4M6. There were no meningeal signs of irritation. Fundus examination was normal. Complete blood count and electrolytes including blood sugar done at admission were non-contributory. Lumbar puncture revealed acellular cerebrospinal fluid (CSF) with normal sugar and protein. Later the CSF culture also came sterile. MRI brain revealed ill-defined asymmetrical, non-enhancing, hyper-intense lesions on T2/FLAIR images involving the subcortical and deep white matter of bilateral frontal, parietal, and temporal lobes (Figure 1). Based on these radiological findings and clinical evidence of encephalopathy and motor symptoms, a presumptive diagnosis of acute disseminated encephalomyelitis (ADEM) was made, and pulse therapy with high dose intravenous (IV) methylprednisolone was given. The patient showed improvement in sensorium (GCS 15/15), jerky movements became passive and was discharged on tapering doses of oral steroids.
 Figure 1: MRI brain performed at six weeks of illness with (a) T1, (b) T2, (c) sagittal, and (d) FLAIR axial sections depicting multiple small discrete sub-cortical white matter lesions (white arrows).
Figure 1: MRI brain performed at six weeks of illness with (a) T1, (b) T2, (c) sagittal, and (d) FLAIR axial sections depicting multiple small discrete sub-cortical white matter lesions (white arrows).
Fifteen days after the discharge, the patient presented with left focal onset motor seizures with impaired awareness, 2–3 episodes per day, each lasting for about 30 seconds, followed by normal consciousness and a history of falling on the left side while walking with the decreased movement of left upper and lower limbs since last 5 days. GCS was E2V2M4 and the fundus examination was normal. Left hemiparesis was evident with the right side normal on examination. The left upper and lower limb was spastic with power 2/5 in the distal muscle groups and 3/5 in the proximal muscle groups with brisk deep tendon reflexes and extensor plantar on the left side. The child was intubated for airway protection in view of poor sensorium. Antiepileptics (IV phenytoin and valproate) were started and repeat pulse therapy with steroids was given. MRI was repeated after the stabilization of the child. It revealed diffuse gyral swelling with T2/FLAIR hyperintensities with diffusion restriction in the frontal, parietal, temporal, and occipital lobes bilaterally. The right external capsule was also involved. Small hyperintensities on T2/FLAIR in the deep white matter of bilateral frontal lobes and periventricular regions without diffusion restriction suggestive of ischemic infarcts were also noted (Figure 2).
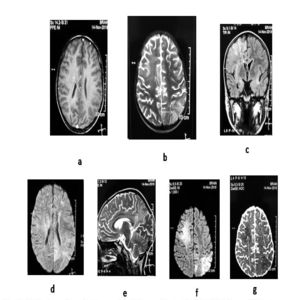 Figure 2: MRI brain performed at ten weeks of illness with (a) axial T1, (b) axial T2, (c) coronal, and (d) sagittal sections, (e) FLAIR, (f) DWI, and (g) ADC depicting multiple infarcts along with sub-cortical white matter lesions (white arrows).
Figure 2: MRI brain performed at ten weeks of illness with (a) axial T1, (b) axial T2, (c) coronal, and (d) sagittal sections, (e) FLAIR, (f) DWI, and (g) ADC depicting multiple infarcts along with sub-cortical white matter lesions (white arrows).
Possibilities of small vessel vasculitis, metabolic encephalopathy, viral encephalitis, and autoimmune encephalitis were considered, and the child was started on a metabolic regime and received IV immunoglobulin after sending a metabolic and autoimmune panel. Injection acyclovir was also started IV. Given the history of myoclonic jerks and progression into seizures and encephalopathy in an unimmunized child possibility of SSPE was also considered. However, the mother denied remembering any history suggestive of measles infection in early childhood. Electroencephalogram (EEG) revealed non-specific slowing but no periodic complexes. Lumbar puncture was repeated and CSF analysis revealed a high titre (> 1:625) for IgG measles which confirmed the diagnosis of SSPE. CSF for HSV PCR was negative. Supportive treatment was continued in the form of mechanical ventilation, seizure control, and anti-raised intracranial pressure measures.
Despite all efforts, his condition continued to deteriorate, and he suffered multiple cardiac arrests and sadly demised. An autopsy could not be performed in the absence of consent.
SSPE is caused by the persistence of the mutated/defective MeV in the central nervous system after primary infection. Various reasons for this persistence of infection have been postulated, including mutation in the virion, defect in the virus, impaired host immune system, and genetic susceptibility of the host [4]. Chronic infection of the central nervous tissue by this virion leads to invariably fatal encephalitis after a variable interval of time known as the latent period. In most cases, the period of latency is three to eight years, while it can vary from one to twelve years [1, 5]. The early presenting features of SSPE include cognitive disturbances in a child with myoclonic jerks, ataxia, and seizures. However, tremors, dystonias, headaches, hemiparkinsonism, and hallucinations have also been described. The disease progresses through various stages, categorized by Jabbour et al. [6]. The disease has a fatal outcome in most cases over a period of 1 to 3 years [1]. Fulminant course with a total span of three months to one year between stage I-II to stage IV (vegetative and death) have also been reported [3]. This catastrophic course has been attributed to the nature of the mutation, the immune status of the host, and sometimes even the use of steroids [4].
Diagnosis can be based on clinical, electrophysiological, immunological, and pathological evidence, but all of these may not be easily available. That is why imaging techniques like MRI are gaining prominence in diagnosing the disease. MRI is fast becoming the initial investigation, although variability in the MRI findings is more of a rule than an exception [7]. The MRI changes in SSPE are non-specific, most commonly signal changes are noted in white as well as grey matters of the parietal and occipital lobes in the form of hyperintensities on T2-weighted and FLAIR sequences. It is not common to see edema, mass effect, or contrast enhancement. Despite the advances, diagnoses ranging from ADEM, cerebral gliomatosis to Rasmussen’s encephalitis were made erroneously in children suffering from SSPE [8–10]. Our patient was also misdiagnosed as ADEM on the first MRI for which he received steroids which have been described to aggravate the condition in some cases [4]. Gyral swelling has also been reported rarely in the literature [7]. The MRI done at six weeks of illness showed diffuse gyral swelling along with the findings suggestive of acute to sub-acute white matter involvement in the form of ischemic infarcts. These findings are not typical of SSPE. The inability to elaborate a confirmatory history of measles in early childhood from the mother and the absence of typical findings on MRI compromised the chances of timely and correct diagnosis in our patient.
In the setting of low health awareness and general illiteracy, the history of precedent measles infection may not always be forthcoming. A high index of suspicion is important in cases with SSPE based on clinical presentation. Confirming the immunization status is crucial. MRI can help in making a diagnosis, but all the possible changes encompassing all stages and their correlation with severity are yet to be explored.
