Background: Diagnosis of rare diseases or disorders is understandably challenging because it is unfeasible for practicing physicians to make themselves familiar with hundreds of rare diseases. The diagnosis of rare inherited metabolic syndromes such as mucopolysaccharidoses is additionally complicated by the lack of confirmatory sophisticated laboratory tests in many areas of the world. In the more developed countries, the diagnosis of mucopolysaccharidoses depends on urine tests for excessive mucopolysaccharides and enzyme assays. However, these tests are not easily accessible in countries like Iraq, and the diagnosis has to rely on clinical and radiological findings.
Case Report: The clinical and radiologic diagnosis of an Iraqi patient with Maroteaux-Lamy syndrome is described. The clinical diagnosis of the girl with early-onset mucopolysaccharidosis and the abnormalities were recognizable before the age of two. The clinical and radiologic diagnosis was Maroteaux-Lamy syndrome because of the absence of mental retardation and the presence of hepatosplenomegaly.
Conclusion: Clinical diagnosis of rare metabolic syndromes like mucopolysaccharidoses requires magnificent clinical skills and huge experience because of the similarity between various types of mucopolysaccharidoses.
Maroteaux-Lamy syndrome, diagnostic challenge, Iraq
Diagnosis of rare diseases or disorders is understandably challenging because it is unfeasible for practicing physicians to make themselves familiar with hundreds of rare diseases. The diagnosis of rare inherited metabolic syndromes such as mucopolysaccharidoses is additionally complicated by the lack of confirmatory sophisticated laboratory tests in many areas of the world. In the more developed countries, the diagnosis of mucopolysaccharidoses depends on urine tests for excessive mucopolysaccharides and enzyme assays. However, these tests are not easily accessible in countries like Iraq, and the diagnosis has to rely on clinical and radiological findings [1–3].
The clinical and radiological diagnosis of an Iraqi patient with Maroteaux-Lamy syndrome is described. The patient was a five and half years old girl who is known to have a metabolic storage disease. The girl was seen during April, 2016 as she was suffering from breathing problem that is mostly attributed to recurrent respiratory infections and obstructive airway disease. Her parents were related and her older sister had the same condition who died before the age of 10 years from cardiac problem and was blind before death, but she didn’t experience intellectual deterioration.
At the age of one and half year (April, 2012), it was clear that this patient had coarse facial features and abdominal enlargement. Her motor and mental development was considered normal during that time.
Metabolic screening tests were performed and included urinary tests for homocysteine, cystine, phenylketonuria, homogentisic acid, and ketoacids; they were all negative. There was no glucosuria and Benedict’s test for reducing substances was also negative. Urine chromatography for amino acids showed normal findings, but serum chromatography was positive for methionine.
During April, 2012 her heart was also evaluated with echocardiography which showed persistent thymus which may give the impression of cardiomegaly. The figure shows the coarse facial features of the child at the age of five and half years during April, 2016 and shows the abdominal enlargement caused by hepatosplenomegaly with spleen reaching the umbilicus (Figure 1). The child had thick eyebrows, a depressed nasal bridge, thick lips, and large mouth and tongue (macroglossia). The child also had very stiff joints with contractures at the knees which made it difficult for her to walk. Her mental and motor development was normal. Neurologic examination was normal with no evidence of spasticity and she had no hearing impairment.

Figure 1: The girl had coarse facial features and abdominal enlargement caused by hepatosplenomegaly with spleen reaching the umbilicus.
Bone radiographs showed evidence of severe skeletal dysplasia with bone enlargement, irregular shape, and bowing of bones (Figure 2).
The clinical diagnosis of this girl with early onset mucopolysaccharidosis and the abnormalities were recognizable before the age of two. The clinical and radiologic diagnosis was Maroteaux-Lamy syndrome because of the absence of mental retardation and the presence of hepatosplenomegaly.
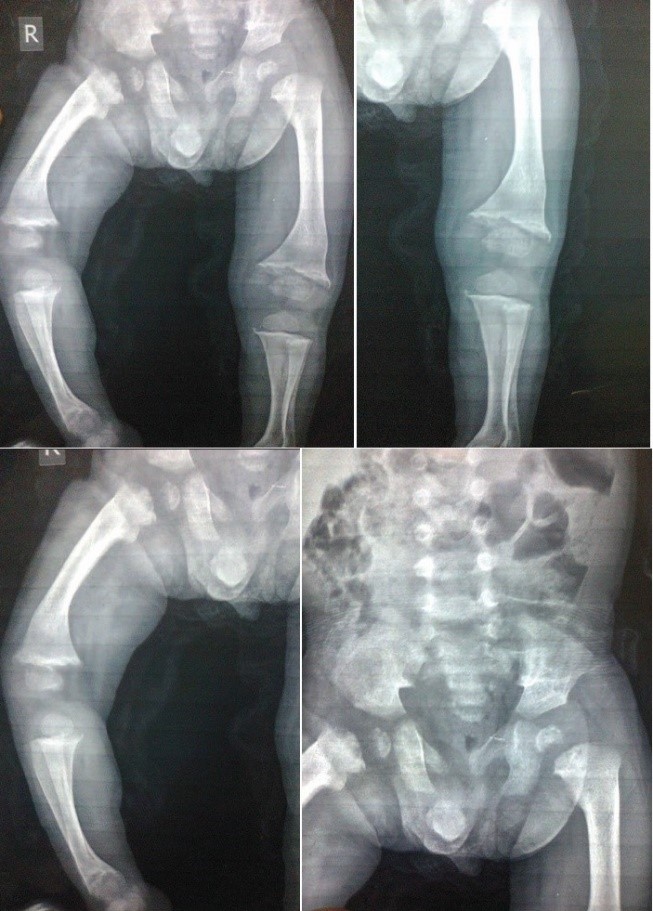
Figure 2: Bone radiographs showing evidence of severe skeletal dysplasia with bone enlargement, irregular shape, and bowing of bones.
Mucopolysaccharidoses syndromes were first described by Gertrud Hurler, a German pediatrician in 1917 and by Charles A Hunter in 1919 as syndromes of chondrodystrophic changes in the skeleton, corneal opacities, hepatosplenomegaly and mental retardation. In 1936, Ellis et al. called the mucopolysaccharidoses syndromes gargoylism [3–6].
Hurler syndrome is the severe form of this disorder with death usually occurring by the age of 10 years. The syndrome is characterized by coarse facial features, short stature, skeletal deformities, joint stiffness, and mental retardation with onset of disease usually between 2–4 years of age. Scheie syndrome is the mild form of mucopolysaccharidoses. Onset of significant symptoms are usually recognized at about the age of six years with diagnosis generally made after ten years. Scheie Syndrome is associated with stiff joints, aortic valve abnormalities, corneal clouding, degeneration of the retina and airway obstruction. However, patients with Scheie syndrome have normal intelligence and significant mental retardation doesn’t occur.
Hurler-Scheie syndrome is a clinical phenotype that is intermediate between Hurler and Scheie syndromes. The somatic involvement is progressive, but with little or no intellectual dysfunction. Symptoms are commonly observed between three and eight years of age, and survival to adulthood is common. Cardiac involvement and upper airway obstruction contribute to clinical mortality [3, 7–10].
Morquio syndrome is associated with a skeletal dysplasia that is distinct from other mucopolysaccharidoses with a short trunk and neck, fine corneal deposits and the absence of mental retardation. The appearance of genu valgum and waddling gait with a tendency to fall are some of the early symptoms of Morquio syndrome. Coarsening of facial features is less significant than in other types of mucopolysaccharidoses [3].
Hurler and Hunter syndromes are characterized by significant mental retardation and cannot be included in the differential diagnosis of the girl with mucopolysaccharidosis without significant mental retardation. Hunter syndrome has an X-linked recessive inheritance, and occur in males [3–6].
Coarsening of facial features and hepatomegaly are less significant in Morquio syndrome than in other types of mucopolysaccharidosis. Morquio syndrome cannot be considered in the differential diagnosis of the girl with mucopolysaccharidosis with hepatosplenomegaly and significant coarsening of facial features. Scheie and Hurler-Scheie syndromes can occur in girls without significant mental retardation but they are not associated with early onset and are generally not diagnosed before the age of two years [3–10].
Maroteaux-Lamy syndrome commonly has early-onset of symptoms and the diagnosis earlier than the age of two. It is characterized by coarse facial features, joint stiffness, valvular heart abnormality, and dysostosis multiplex, and the lack of mental retardation. Mild and intermediate forms of Maroteaux-Lamy syndrome are very similar to Scheie syndrome [3, 11–13].
Maroteaux-Lamy syndrome was also called Polydystrophic dwarfism, Mucopolysaccharidosis VI (MPS 6), and arylsulfatase B deficiency (ARSB deficiency). It is an autosomal recessive type of mucopolysaccharidosis which is a lysosomal storage disorder that was first described in 1963 by French physicians Pierre Maroteaux and his mentor Maurice Emil Joseph Lamy. They described the condition as a novel dysostosis associated with increased urinary excretion of chondroitin sulfate.
Children with Maroteaux-Lamy syndrome are generally not mentally retarded but have many of the physical symptoms of Hurler syndrome. Maroteaux-Lamy syndrome has a variable severity of manifestations which usually includes short stature, hepatosplenomegaly, dysostosis multiplex, stiff joints, corneal clouding, cardiac abnormalities, and facial dysmorphism. Affected children will gradually develop a shortening of the trunk, crouched stance, restricted joint movement, a protruding abdomen and forward-curving spine. The skeletal changes are progressive and restrict movement. Affected children may have umbilical hernia or inguinal hernia. Most children with this disorder develop cardiac abnormality mostly in the form of valvular dysfunction [3, 11–13].
The recent availability of treatment with enzyme replacement for some types of mucopolysaccharidoses made the clinical diagnosis of such disorders of practical value as it may give the patients the chance to have the new treatment in another country when this is possible [3].
Clinical diagnosis of rare metabolic syndromes like mucopolysaccharidoses requires magnificent clinical skills and huge experience because of the similarity between various types of mucopolysaccharidoses.
The author would like to express his gratitude towards the parents of the patient who accepted to publish her photos for the case report.
